Reposted from Dr. Judith Curry’s Climate Etc.
Posted on December 29, 2020 by niclewis
By Nic Lewis
Key points
- A new variant, B.1.1.7, of the SARS-CoV-2 virus has recently spread rapidly in England
- The public health agency’s best estimate of B.1.1.7’s weekly growth rate advantage is 1.51x
- They mis-convert this in a reproduction number ratio of 1.47; converting appropriately gives a ratio of 1.25
- Confident claims by the UK government scientific advisers that the higher growth of B.1.1.7 is due to increased transmissibility are misplaced; it could be partly of wholly due to other factors
- 1.1.7 has not shown a greater growth rate advantage than two previous variants did, both of which are now thought to have no greater transmissibility than previously existing variants
- There is little evidence that B.1.1.7 is more virulent, or likely to be resistant to existing vaccines
Introduction
The apparent rapid growth in England of a new variant of the SARS-CoV-2 virus that causes COVID-19 has led to dire warnings by those advising the UK government. Their advice suggested only that the new variant was more transmissible (more infective), not that it was more virulent (causes more serious illness). Nevertheless, it resulted in swift (many would say panicky) actions first by the UK government and then by governments of many other countries. The UK government imposed further restrictions on people’s freedom to mix and to move, while other countries banned travellers from the UK. Many millions of people in the UK had to cancel their plans for the Christmas holiday at very short notice, in addition to having their freedoms further curtailed thereafter. In this article I examine to what extent the advice that led to these damaging government actions was justified.
The new strain, B.1.1.7, and its spread in the UK
The UK government agency Public Health England (PHE) termed the new variant VUI-202012/01, and now VUC-202012/01, but I shall refer to it by the scientific name given to its lineage, B.1.1.7 (Rambaut et al.)[1], or just as “the new variant”. The lineage involves 8 amino acid changes (6 mutations and 2 deletions)[2] in the gene for the important spike protein, along with 9 amino acid changes[3] in genes for other proteins. The lineage has sometimes been referred to just by the name of the best known mutation it possesses, N501Y, but doing so is to be avoided as there are other variants that also have this mutation.
Rambaut et al. have this to say about the new lineage:
The B.1.1.7 lineage carries a larger than usual number of virus genetic changes. The accrual of 14 lineage-specific amino acid replacements prior to its detection is, to date, unprecedented in the global virus genomic data for the COVID-19 pandemic.
They also identify three of the mutations in particular (including N501Y) as being suspected of having potential biological effects.
B.1.1.7 was first detected in SARS-CoV-2 sequenced from a sample collected in south-east England on 20 September 2020, since when the cluster of cases has grown rapidly and spread to other locations. The UK sequences many more SARS-CoV-2 genomes than any other nation, and more than the rest of Europe combined, so the fact that B.1.17 was first detected in the UK does not necessarily imply that it originated there. The lineage has also been detected in several other countries and may well now be widespread.
Growth of the B.1.1.7 lineage in the UK can be tracked in sequencing data uploaded to GISAID. I used the COVID-CG processing facility[4] to select each day’s sequences with all eight B1.1.7 spike gene mutations.[5] As the daily data were noisy and few sequences were dated after 12 December 2020, I took 7-day moving averages, centred up to 9 December. Figure 1 shows the resulting proportion of all UK sequences represented by the B.1.1.7 lineage since its first emergence. It should be noted that the proportion of non-B.1.1.7 sequences represented by the areas in which B.1.1.7 first grew to prominence may have increased over time, resulting in the growth shown overstating how fast it grew in individual areas or in the UK as a whole.

Figure 1. The proportion of all SARS-CoV-2 genomes sequenced in the UK made up by the B.1.1.7 lineage
The higher growth rate of B.1.1.7
A PHE report published on 21 December 2020[6] presents epidemiological evidence about the growth rate of B.1.1.7 relative to non-B.1.1.7 lineages. By using a proxy marker for B.1.1.7[7] they were able to utilise data from a significant proportion of the UK ‘pillar 2’ testing programme. Doing so provided a much larger dataset than that of sequenced SARS-CoV-2 genomes, and enabled stratification of weekly data for each of 42 NHS “STP” areas. Figure 2 reproduces Figure 1 of the PHE document, which shows the multiplicative advantage in weekly growth rates of B.1.1.7 cases (the ratio of B.1.1.7 to non-B.1.1.7 week t+1 cases divided by week t cases). The x-axis is for the B.1.1.7 proxy, S gene test negative. The week stated is the base week, so the yellow points reflect ratios of week 49 (week ending 5 December) cases to week 48 cases.

Figure 2. Empirical data analysis of the multiplicative advantage in weekly growth rates. Each point represents the ratio of weekly growth rates between B.1.17 [VOC] and non-B.1.1.7 for an NHS England STP area and week, based on the pillar 2 data shown in Figure S1 of the PHE report. Colours and shapes differentiate calendar weeks. Numbers above 1 show a multiplicative advantage. The blue line represents the mean value for a particular frequency, and the grey lines the 95% envelope. Scatter at low frequencies largely reflects statistical noise due to low counts.
When the new variant represents a small proportion of total cases (under ~ 25%, say), the proxy used is less satisfactory, and there is also a lot of scatter. Nevertheless, the variant’s proxy-based mean multiplicative advantage (ratio) in weekly growth is remarkably independent of its relative prevalence. That supports PHE’s methodology, although the week 48 data suggests that the multiplicative advantage might decline once the variant makes up more than ~25% of the total cases. PHE compute from this data a mean multiplicative advantage in weekly growth of 1.51 for B.1.1.7. By assuming a fixed generation interval of 6.5 days, they convert this into a reproduction number (Rt) multiplicative advantage of 1.47 for B.1.1.7 relative to other variants,.
PHE also estimated the effect of B.1.1.7 on Rt using genomic (sequencing) data for the same areas and weeks. They estimated an additive effect on Rt of 0.57, or 0.74 when the effect was allowed to vary between areas. PHE also estimated the effect on Rt using the PCR test S gene proxy data, adjusted for specificity (which is poor when the S gene negative proportion is low). Their estimates of the additive effect on Rt using that data were 0.52, or 0.60 when the effect was allowed to vary between areas. Using a Bayesian regression model their estimate of the effect was 0.56. However, since any biological difference in infectivity would be expected to cause a multiplicative effect on Rt, and Rt was varying during the analysis period, an estimated additive effect on Rt is less useful and also liable to be less accurate than a multiplicative estimate. In addition all these estimates involve more complicated statistical models, further assumptions and estimates of other variables. I therefore prefer their estimated multiplicative advantage of 1.51 (for weekly growth, prior to conversion to Rt scale), which is directly derived from underlying data. This is equivalent to a logarithmic daily growth rate advantage of 0.059.
Other evidence regarding the faster growth of B.1.1.7
A meeting of the NERVTAG[8] committee – which advises the government on the threat posed by new and emerging respiratory viruses – on the new variant took place on the 18 December 2020. The minutes[9] refer to an estimate from genomic data of a growth rate 71% higher than other variants; none of the documents that was considered by the committee contained such an estimate. It appears from the minutes of a subsequent meeting on 21 December 2020[10] that this was one of several undocumented estimates from NERVTAG member Professor Neil Ferguson of Imperial College. An alternative regression estimate that he apparently presented indicated that lineage B.1.1.7 had a Rt 0.39 higher than non-variant lineages from early November to early December. This is presumably an additive effect estimate, and is noticeably lower than PHE’s estimates using much the same method. Two other estimates stated in the minutes to be from Professor Ferguson both appear to actually be slightly misstated versions of the PHE estimate of a multiplicative Rt advantage of 1.47 for B.1.1.7.
The minutes of a further NERVTAG meeting the 21 December also mention a London School of Hygiene and Tropical Medicine estimate that B.1.1.7 was 56% more transmissible (a multiplicative advantage of 1.56 in Rt value). This estimate is documented in a preprint (Davies et al.[11]). The authors use a Subjective Bayesian method to fit a highly complex model with many probabilistic parameters, some fixed and others estimated. This is a far from robust, and quite possibly inherently biased, method of estimating the relative transmission rate. Moreover, they appear to use less informative data, broken down geographically only by the 7 NHS regions, not (as PHE used) by the 42 NHS STP areas. The use of less informative data implies that, even if they had employed a robust method, their estimates would be expected to be inherently less reliable than the PHE estimate. The uncertainty ranges of their estimates – which include a 99%+ confidence interval of 1.49x to 1.57x for the South East[12] region – appear to be quite unrealistically narrow, given the substantial uncertainties that exist. That casts further doubt on the realism of their estimates.
Finally, the NERVTAG 21 December meeting also considered a University of Edinburgh (Andrew Rambaut) phylogenetic estimate based on genetic sequences from Kent and London, that Rt was 1.57 or 1.72 depending on the time window used. Since no comparative Rt estimate for non-B.1.1.7 variants is mentioned in the meeting minutes, it is not possible to infer from this an estimate of the relative transmission rate of the new variant.
I conclude that the other evidence considered by NERVTAG is less robust and less useful than the PHE estimate of a multiplicative advantage in weekly growth of 1.51.
Why the faster growth of B.1.1.7 need not be due to increased transmissibility
While the evidence that the B.1.1.7 lineage has grown faster than other lineages in England over the two months or so to early/mid-December seems robust, one cannot infer biological properties of a virus from limited epidemiological data only. The apparent rapid spread of this new variant might be due to founder effects and super-spreader events rather than, or in addition to, increased transmissibility (higher infectivity).
It is instructive in this regard to consider two other lineages/variants that also had a period of exceptionally fast growth and, in one case, came to be totally dominant in most countries.
The G clade: spike gene mutation D614G
The D614G mutation arose early in the epidemic, emerging in Europe in February, and the G614 variant undoubtedly spread faster in most locations than D614. In very many countries, areas and cities it went from representing a minority of infections to being the dominant variant within a period of month or so. Since July 2020 it has accounted for approaching 100% of new infections in most countries and in all continents.
In the light of D614G becoming and remaining so dominant, it is unsurprising that a paper in August (Korber et al.[13]) argued that the D614G mutation increases transmissibility, citing several pieces of evidence:
- the consistency of increase [in frequency of G614] across geographic regions.
- the D614 form did not persist in many locations where the G614 form was introduced into the ongoing well-established D614 epidemics, as would be expected if the two forms were equally likely to propagate.
- the increase in G614 frequency often continued well after national stay-at-home orders [lockdowns] were in place, when serial reseeding from travellers was likely to be reduced significantly.
In addition to that epidemiological evidence, the authors also noted that increased transmissibility of G614 was consistent with other studies that suggested associations with increased infectivity in vitro[14] [15], and with their own finding of an association with higher viral loads in vivo. Moreover, another paper (Li, Q et al[16]) reported higher antigenicity for G614.
Most of Korber’s arguments are also fairly applicable to evidence suggesting that B.1.1.7 may be more transmissible. However, a more recent paper in Nature (van Dorp et al.[17]) found “no evidence for significantly more transmissible lineages of SARS-CoV-2 due to recurrent mutations”, including D614G (B.1.1.7 had not been identified by the end of the study period). This shows that, even after a new strain has become dominant, conclusions about its relative transmissibility drawn from epidemiological and indirect biological evidence may turn out to be wrong.
The 20A.EU1variant: spike mutation A222V
The 20A.EU1 variant, which involves spike gene mutation A222V, emerged in Spain in early summer 2020. It rapidly spread to other European countries, where it typically grew faster than non-20A.EU1 variants. Figure 3 plots the logarithmic daily growth rate of sequences with the A222V mutation in the UK, relative to those without it, over the two months to mid-September. Over that period the ratio of A222V to non-A222V new sequences grew from under 0.02 to 0.67. The mean logarithmic daily growth rate was 0.061 – a weekly multiplicative advantage of 1.53 – with essentially no trend. That multiplicative advantage is effectively identical to the 1.51 estimate by PHE for B.1.1.7 using data from mid-October to mid-December.

Figure 3. The logarithmic daily growth rate of the 7-day moving average of new sequences with the A222V mutation in the UK, relative to those without it, over the two months to 12 September 2020.
However, in the autumn the relative frequency of new A222V sequences stopped increasing in a number of countries, without – as D614G did – achieving total and continuous dominance (Figure 4). In the UK the A20.EU1 variant reached some 70% of all new sequences by the end of October, but it has since declined in relative frequency, as it has also done in Belgium, Germany and Switzerland.

Figure 4. The proportion during 2020 of weekly new SARS-CoV-2 sequences in ten European countries that have the A222V mutation (implying they are the A20.EU1 variant)
Notwithstanding the rapid growth of the 20A.EU1 variant in many European countries during the summer and/or autumn, a November 2020 preprint paper about 20A.EU1 concluded: “We find no evidence of increased transmissibility of this variant, but instead demonstrate how rising incidence in Spain, resumption of travel across Europe, and lack of effective screening and containment may explain the variant’s success”.[18]
Overestimation of effect on Rt of possible increased transmissibility
Supposing that the higher growth rate to date of the B.1.1.7 lineage were all due to higher transmissibility, what effect would this have on the current reproduction number, Rt? That will depend on what Rt is and on the mean generation interval and its probability distribution. The longer the generation interval, the higher Rt required to produce a given growth rate. In the 21 December PHE publication, their estimate of the multiplicative advantage in weekly growth rate (of 1.51) is converted to a multiplicative advantage in Rt of 1.47 by assuming a fixed generation interval of 6.5 days: 1.47 = 1.51^(6.5/7).
However, PHE’s conversion formula is not justified, for two reasons:
- the generation interval is not fixed; and
- recent estimates of the mean generation interval are well below 6.5 days.
Most estimates of the generation interval (the period from one person being infected to them infecting another person) are in fact estimates of the serial interval (the period from the symptom onset in one person to symptom onset in a person they infect), since the time of infection is not observable. The generation interval can be validly estimated by combining probabilistic estimates of the serial interval and the incubation period (from infection to symptom onset). However, simply treating a probabilistic serial interval estimate as representing the generation interval distribution, as is typically done, is unsatisfactory.
PHE give no source for their assumption of a 6.5 days generation interval, but they could be following the Imperial College team, who used (in Flaxman et al.[19]) a generation interval with a 6.5 days mean, stating it was estimated by Bi et al.[20]. In fact, Bi et al. estimated the serial interval, not the generation interval, and fitted a gamma probability distribution with a mean of 6.3 days. However, their data included cases where the infecting individual did not isolate from others until long after symptom onset. Bi et al. found that if the infected individual was isolated less than three days after symptom onset, which would normally be the case in the UK now, the average serial interval was only 3.6 days.
Knight and Mishra (2020)[21] show that, to avoid overestimating the serial interval, it needs to be fitted to a probability distribution that, unlike the gamma distribution used by Bi et al., permits negative values (which are observed in a non-negligible proportion of cases). They consider a number of estimates examined in a review article of the incubation period and the serial interval, selecting the only serial interval estimate based on a negative-permitting probability distribution that had a large sample size (nearly ten times as large as that in Bi et al.), and the incubation period estimate that was based on the largest sample. Their resulting generation interval estimate has a mean of 3.99 days and a standard deviation of 2.96 days.[22]
Davies et al. say that their complex Bayesian model, which estimated a multiplicative advantage of 1.56 in Rt value using a fairly long generation interval, fitted less well when they used a shorter interval. However, it seems probable that the main reason they obtain a poor fit to the relative growth in the new variant during lockdown with a shorter generation interval is that their model greatly overestimates the effect of the November lockdown. [23]
Using the Knight and Mishra estimated distribution for the generation interval, and the correct conversion formula,[24] the PHE estimate of the B.1.1.7 lineage’s multiplicative advantage in weekly growth rate of 1.51 corresponds to a multiplicative advantage in Rt of 1.25.[25] That is only about half the multiplicative advantage of 1.47 estimated by PHE.[26] It implies that less extensive and severe measures would be required to prevent exponential growth of infections given the emergence of B.1.1.7 than is implied by the PHE estimate of a 1.47 multiplicative advantage in Rt, even if the observed multiplicative advantage in weekly growth rate of B.1.1.7 to mid-December is entirely caused by it being more transmissible.
The new South African variant
A new SARS-CoV-2 lineage that also involves a N501Y spike gene mutation, and a number of other mutations (differing from those in B.1.1.7), has recently emerged in South Africa, as described by Tegally et al.[27], who term it 501Y.V2. They say that genomic data, showing the rapid displacement of other lineages, suggest that this lineage may be associated with increased transmissibility. However, the limited evidence available so far is insufficient to justify the alarmist comments from the UK government health minister, that “This new variant is highly concerning because it is yet more transmissible and it appears to have mutated further than the new variant discovered in the UK”.[28] As a professor of molecular virology at the University of Nottingham stated, the mutation involved has been seen before, we have no idea whether it impacts on virus transmissibility, and we should avoid panicking at this point.[29]
Conclusions
The G clade and 20A.EU1 examples illustrate that apparently strong epidemiological evidence of a higher growth rate of a new variant over considerable period, even where it leads to apparently permanent dominance, and notwithstanding it being accompanied by evidence suggesting that the variant is associated with higher viral loads, does not enable a valid conclusion that the variant has higher transmissibility than existing variants. Such evidence may be suggestive of higher transmissibility, but it does not reliably demonstrate it.
Despite this, the NERVTAG committee concluded with moderate confidence on 18 December that the new variant “demonstrates a substantial increase in transmissibility compared to other variants” and at their 21 December meeting went further and expressed high confidence that “B.1.1.7 can spread faster than other SARS-CoV-2 virus variants currently circulating in the UK”. While admitting that the underlying cause of faster spread was unclear, the causative factors that they suggested related exclusively to higher transmissibility. It is not surprising, given that NERVTAG’s confident conclusions are not justified by the facts, that a number of experts in the UK and other countries have disputed them[30] or expressed contrary views,[31] [32] [33] Unsurprisingly, the mainstream media are reporting, incorrectly, that B.1.1.7 has been proven to possess substantially increased transmissibility.
I have argued that the estimate by PHE of a multiplicative weekly growth advantage of 1.51 for B.1.1.7 is, for several reasons, more robust and accurate than the other available estimates. I have shown that, even if the higher growth to date of B.1.1.7 were due entirely to increased transmissibility, it would correspond to a multiplicative advantage in Rt of only 1.25, half as high an advantage as calculated by PHE using an inappropriate conversion formula.
There is no evidence to date that B.1.1.7 is any more any virulent than existing strains, nor that it will be resistant to the vaccines that have been developed. Expert opinion appears to be that neither of these are likely to be the case.[34]
These findings imply that B.1.1.7 does not currently appear to represent a serious increase in the menace posed by SARS-CoV-2, even in the worst case that its higher observed growth rate is entirely due to increased transmissibility. In the best case, its higher growth rate will turn out not to spring to any extent from increased transmissibility, as now appears to be the case with the G clade and 20A.EU1 variant.
Accordingly, it is difficult to see that imposing drastic measures to slow transmission, further reducing economic activity, social activity and peoples freedom, are justified by the current evidence regarding the emerging B.1.1.7 lineage.
However, as further evidence becomes available it could strengthen, or weaken, the case that the emergence of B.1.1.7 represents a serious development. It is important that the UK authorities start to release, on a daily basis and at local authority area or finer level, all available data on cases of the new strain, as indicated by the ‘S gene negative’ proxy and any other method. At present they are keeping this information non-public, which makes it impossible for independent researchers properly to assess on a timely basis, and if necessary challenge, what may be mistaken conclusions. Moreover, it is highly desirable that no SARS-CoV-2 or COVID-19 related report or study should hereafter be considered by the government or its advisers unless it is accompanied by a link at which all the data used is available.
Nicholas Lewis 29 December 2020
[1] Rambaut, A et al: Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. COVID-19 Genomics Consortium UK, ARTIC network 19 December 2020. https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563
[2] N501Y, A570D, P681H, T716I, S982A and D1118H mutations plus HV69-70 and Y144 deletions
[3] T1001I, A1708D and I2230T mutations plus SGF3675-3677 deletion in the ORF1ab gene; R52I and Y73C mutations plus Q27stop codon in the Orf8 gene; D3L and S235F in the N gene. There are also 6 synonymous (non-amino acid changing) mutations: 5 in ORF1ab (C913T, C5986T, C14676T, C15279T, C16176T), and 1 in the M gene (T26801C).
[4] https://www.covidcg.org/?tab=location# . Data downloaded 26 December 2020.
[5] Only the total number of each day’s sequences with each mutation are available via COVID-CG, but the number of each of the eight spike mutations appearing each day (r >0.999, except 0.991for the HV69-70 deletion, which sometimes occurs in other strains), implying that they have an extremely high co-occurrence. I took the minimum number each day for the eight spike mutations as the count for B.1.1.7 sequences. Incorporating non-spike mutation data appears unnecessary; the match of all B.1.1.7 spike mutations with all ORF1ab B.1.1.7 mutations is almost perfect apart from A1708D, which seems absent in about 1% of cases where all 11 other spike and ORF1ab mutations are present.
[6] Public Health England: Investigation of novel SARS-COV-2 variant – Variant of Concern 202012/01. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/947048/Technical_Briefing_VOC_SH_NJL2_SH2.pdf
[7] S gene negative, N and ORF1ab positive TaqPath PCR test result.
[8] NERVTAG: New and Emerging Respiratory Virus Threats Advisory Group
[9] NERVTAG COVID-19 VUI communication 18122020_final.pdf, available at https://app.box.com/s/3lkcbxepqixkg4mv640dpvvg978ixjtf/folder/111416414559
[10] NERVTAG COVID-19 VOC communication 21122020 final.pdf, available at https://app.box.com/s/3lkcbxepqixkg4mv640dpvvg978ixjtf/folder/111416414559
[11] Davies, NG et al: Estimated transmissibility and severity of novel SARS-CoV-2 Variant of Concern 202012/01 in England. Centre for Mathematical Modelling of Infectious Diseases, London School of Hygiene and Tropical Medicine, updated 23 December 2020. https://cmmid.github.io/topics/covid19/reports/uk-novel-variant/2020_12_23_Transmissibility_and_severity_of_VOC_202012_01_in_England.pdf
[12] Figure1A of Davies et al, rightmost panel.
[13] Korber, B. et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 182, 812.e19–827.e19 (2020). https://doi.org/10.1016/j.cell.2020.06.043, 20 August 2020
[14] Zhang, L. et al. The D614G mutation in the SARS-CoV-2 spike protein educes S1 shedding and increases infectivity. Preprint at https://doi.org/10.1101/2020.06.12.148726, 12 June 2020
[15] Yurkovetskiy, L. et al. Structural and functional analysis of the D614G SARSCoV-2 spike protein variant. Cell 183, 739.e8–751.e8 https://doi.org/10.1016/j.cell.2020.09.032, October 2020
[16] Li, Q. et al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 182, 1284.e9–1294.e https://doi.org/10.1016/j.cell.2020.07.012, September 2020
[17] van Dorp, L et al., No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nature, November 2020. https://doi.org/10.1038/s41467-020-19818-2
[18]Hodcroft, BH et al: Emergence and spread of a SARS-CoV-2 variant through Europe in the summer of 2020. medRxiv 27 November 2020 https://doi.org/10.1101/2020.10.25.20219063
[19] Flaxman, S., Mishra, S., Gandy, A. et al. Estimating the effects of non-pharmaceutical interventions on COVID-19 in Europe. Nature 584, 257–261 (2020). https://doi.org/10.1038/s41586-020-2405-7
[20] Bi, Q. et al. Epidemiology and Transmission of COVID-19 in Shenzhen China: Analysis of 391 cases and 1,286 of their close contacts. medRxiv (2020) https://doi.org/10.1101/2020.03.03.20028423
[21] Knight, J. and Mishra, S.: Estimating effective reproduction number using generation time versus serial interval, with application to COVID-19 in the Greater Toronto Area, Canada. Infectious Disease Modelling 5 (2020) 889e896, November 2020. https://doi.org/10.1016/j.idm.2020.10.009
[22] Knight and Mishra fit their generation interval estimate using a gamma distribution. Unlike the serial interval, the generation interval cannot be negative so a gamma distribution is suitable here.
[23] They say that the poor fitting with a shorter generation interval was because it predicted that the new strain should have decreased in relative frequency during November’s lockdown. As they write: “When Rt < 1 for both variants, a shorter generation time is a selective disadvantage, because infections with this variant decline faster compared to a variant with the same Rt but transmitting on a longer timescale.” However, that is only true if Rt was below 1 during lockdown, whereas their Fig. 1E shows that, in reality, Rt remained at or marginally above 1 during lockdown. That is consistent with the mobility data in Davies et al. Fig.1C, which show little difference between immediately prior to the start and the end of lockdown. An overall Rt of 1 implies that the infections with the more transmissible variant will increase in relative frequency, as occurred, not decrease. Also, the complexity of their model means that the poor fitting could to be partially or wholly due to other causes, such as a long generation interval compensating for another parameter being misestimated, or to the peculiar way in which they represented a shortened generation interval.
[24] Wallinga, J., & Lipsitch, M. (2007). How generation intervals shape the relationship between growth rates and reproductive numbers. Proceedings of the Royal Society B: Biological Sciences, 274(1609), 599-604. https://doi.org/10.1098/rspb.2006.3754 Using their equation 2.9 in conjunction with the gamma distribution moment generating function.
[25] The estimate of the multiplicative advantage assumes that Rt for the other strains is 1.0; the inferred multiplicative advantage is a slowly decreasing function of Rt for the other strains.
[26] The same is approximately true throughout PHE’s 95% confidence interval of for the Rt ratio of 1.34–1.59, which when converted in the same way corresponds to an Rt ratio range of 1.19–1.30.
[27] Tegally, Houriiyah, et al. “Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa.” medRxiv 22 December 2020. https://doi.org/10.1101/2020.12.21.20248640
[28] As quoted in the Guardian, 23 December 2020. https://www.theguardian.com/world/2020/dec/23/south-african-covid-19-variant-has-reached-the-uk-says-matt-hancock
[29] Professor John Ball, as quoted at https://www.sciencemediacentre.org/expert-reaction-to-south-african-variant-of-sars-cov-2-as-mentioned-by-matt-hancock-at-the-downing-street-press-briefing/
[30] Vincent Racaniello, Professor of Immunology, Columbia University. Extensive detailed comments, including that “none of the isolates so far have proven implications for human transmission or pathogenesis, including the latest variant isolated from the UK.” and, concerning the NERVTAG 21 December meeting: “You can’t use epidemiological data to prove a biological effect of a amino acid change in a virus; you have to do experiments to do that. And that’s what they’re doing here. They say, there is an increase in the transmissibility. It must be because of the variant. Well, obviously that’s a flawed argument. That’s not how we do science.” https://www.virology.ws/2020/12/24/sars-cov-2-uk-variant-does-it-matter/ ; transcript at https://dryburgh.com/vincent-racaniello-coronavirus-variant-voc-202012-01/
[31] Dr Julian Tang, Honorary Associate Professor/Clinical Virologist, University of Leicester, said:
“The spread of this new virus variant could be due to many factors. As we saw with the earlier D614G variant – just higher viral loads in clinical diagnostic swabs or in cell culture may not necessarily translate to a more transmissible virus at the population level.
“A higher genomic growth rate in the samples sequenced, may not necessarily mean higher transmissibility, e.g. if there was a rave of several thousand people where this variant was introduced and infected many people mostly in that rave, this may seem very high compared to a lower background of non-variant virus, e.g. in an otherwise prevailing national lockdown.” https://www.sciencemediacentre.org/expert-reaction-to-brief-summary-of-nervtag-opinion-from-the-nervtag-meeting-on-sars-cov-2-variant-under-investigation-vui-202012-01/
[32] Professor Vineet Menachery, University of Texas Medical Branch, said: “So this isn’t the first time we’ve seen variants emerge quickly or begin to dominate the population of viruses that we’re seeing. And so I’m not particularly worried at this moment. There is evidence that it is maybe slightly more transmissible, but we’re not at this point knowing enough about it to really be scared in the sense that it’s a different order of magnitude, that it’s going to be a major threat moving forward.” https://health.wusf.usf.edu/npr-health/2020-12-21/new-coronavirus-variant-found-in-u-k-what-does-it-mean-for-the-world
[33] Dr Nusrat Homaira, Respiratory Epidemiologist, UNSW Sydney. “There is modest evidence suggesting that this new variant of Sars-CoV-2 is more transmissible, and is speculated to be the reason for the recent increase in the number of Covid-19 cases in London, South East, and East of England regions.” https://www.dhakatribune.com/opinion/op-ed/2020/12/24/the-new-variant-of-the-sars-cov-2-what-it-all-means
[34] Dr Julian Tang, Honorary Associate Professor/Clinical Virologist, University of Leicester, said: “We are not seeing any increased virulence (clinical severity) or any gross changes in the S (spike protein) that will reduce vaccine effectiveness – so far.” https://www.sciencemediacentre.org/expert-reaction-to-brief-summary-of-nervtag-opinion-from-the-nervtag-meeting-on-sars-cov-2-variant-under-investigation-vui-202012-01/
Originally posted here, where a pdf copy is also available
Thanks, Nic. Glad to see you’re well and continuing to examine things in realistic ways.
Regards,
Bob
PS: If I don’t comment here again at WUWT within the next couple of days, HAVE A HAPPY AND HEALTHY NEW YEAR, ALL!
Bob
Thanks for the direct analysis and comments, Nic Lewis. The problem for the general public to reasonably understand the Covid-19 pandemic details is two-fold: The issue has become politicized and misused by certain interests, and a large percentage of the general population is not up to studying and understanding the issue. From my limited perspective there are two issues for me to directly consider as I attempt to go about my life, the amount of a viral load I am at what risk of encountering, and the onset of summer (southern hemisphere) as a mitigating effect.
No matter your location on this planet the “Rona will continue to be a menacing hobgoblin by those who control the propaganda press up to and until 80% of the population has been cowed into submission , i.e. accepting conditions imposed by CAG the Central Authoritarian Government (that would be the bureaucratic Deep State here in the United States). The remaining 20% will be forced underground or ‘disappeared” by the oppressing CAG with the presumed dead CoD recorded as COVID..
Also, the absence of a “greenhouse effect” that concentrates viral transmission in eddies, reducing the probability of infection to zero. Not impossible, but improbable.
“The problem for the general public to reasonably understand the Covid-19 pandemic details is two-fold: The issue has become politicized and misused by certain interests, and a large percentage of the general population is not up to studying and understanding the issue.”
A great effort is being made to communicate and get beyond all of the confusion. Check out Pandemics Data and Analytics-PANDA
https://www.facebook.com/PanData19/?fref=mentions&__tn__=K-R
Which foreigners shall we kick today?
China? Russia? How about the Brits?
I am based in Kent in the SE quarter of UK.
We were very lightly affected by the original infection earlier in the year than the rest of the country. Could this be a factor in the increased incidence now.
Yes, through lockdowns and isolation, a community is deprived of community immunity, and the transmission vectors and exponential spread remain viable, awaiting an external introduction of the conatagion.
In your area…absolutely. Lower level of herd immunity. Just needed a spark to go up like tinder.
For the most part in the US, states and even regions hit hard early recovered and have not seen those peaks again, but those that avoided widespread COVID early in the year have been hit hard in recent months.
Sounds reasonable. Is there a study demonstrating this?
They are on a bash, but the pub’s closed….
We Brits are too busy kicking ourselves at the moment, please make an orderly queue
Thank you
Rather than run down a list of nations I prefer to simply refer to them as “others”.
How can ANYONE feel confident in material that Neil Ferguson has gotten his hands on? Apparently the only thing we can be sure of is that panic and fear will be greatly promoted and that dissenting scientific voices will be silenced as much as possible; just as in the climate debate!
Like the UN, the WHO seems to be controlled more by totalitarian and communist entities than by liberal, democratic ones. But even they have had to admit the the CCP inspired lockdown treatments don’t work and are extremely damaging to the poor! Who would have thought that shutting down whole economies would hurt those with little or no money!?
We may have finally found a way to identify and isolate the sociopaths in our midst; it seems they are concentrated in our ruling class elites now where they can practice the fascistic control they have always dreamed of!
I’ve said this many times, how the hell is Ferguson still spreading his doom-laden crap after 20 years of zero successful predictions on the spread of disease?
The climate predictions have been right exactly 0 times over the past several decades, and yet these are still parroted by the media as gospel truth. The message has the desired result, which means the truth does not matter.
If the CoVid measurement method were used to measure snow it would look like the left frame of the picture below & make no reference to the right frame picture.
The ambiguity of science, which is, with cause, a philosophy and practice constrained to a limited frame of reference, but through inference, sincerely held beliefs, and capital or redistributive investments, can imagine a universe, a narrow perspective of climate, a reduction of human life, etc.
The question should be, “Why the hell_o is ANYONE of sound mind, that doesn’t belong to the sociopathic globalist cult, listening to, let alone seeking the advice of, Neil Ferguson!?” In how many ways does he have to be shown and PROVEN to be a Fraud-For-Hire? Gates has him on speed-dial, and Ferguson does his bidding, as Gates is no doubt the highest bidder available.
In Germany they found the new variant in a sample taken in mid November.
This misses the political point.
The UK Government was very keen to have Christmas be “fun” as promised. To provide confidence for the release from restrictions over Christmas they refused to follow the scientific advice and shutdown everything, back at Half-Term. It was called the “Circuit-Breaker”.
The Government was tripped up when the Opposition called for the scientific advice to be followed. Suddenly all the new pressure on the NHS and the additional deaths were clearly the fault of the PM’s misjudgement. The Leader of the Opposition was clearly right.
But a new and more infectious variant is a convenient scapegoat. The PM couldn’t have known about this.
He can get away with it politically – so long as the new variant is hyped up as a whole new challenge.
But please will everyone note that the same advice about space and hand-washing is in place. Nothing has actually changed.
Ah … pressure on the NHS?
https://t.co/dBnf9WqHKq
An empty hospital filmed on a mobile phone (and the lady gets arrested) – the emergency Covid units being disbanded… The media screeching… lock down! and the bureaucrats juggling “tiers”…. to save the NHS…
It doesn’t add up.
Public Health England are doing remarkably well for an outfit that were purportedly euthanised as simply “not fit for purpose” some 4 months ago because they lied (repeatedly and wantonly) about manipulating death statistics and fouled up the testing + it’s a bureaucracy stuffed with failed medics and dimwit managers who’ve got to the top table and by’eck they’re not going to leave without a struggle. PHE were ejected from the NHS by actual clinicians who’d run out of patience with their damaging, stupid antics and they are planning an empire across the land run from a £450 million palace complex next to the M25 motorway at Harlow in Essex.
That the assertions about the “new strain” don’t stand up should come as no surprise to anybody who’s watched PHE for a few years. There is an unhealthy relationship between PHE and the UK’s MSM – who aren’ bright enough for the most part to come up with the scary shit they’ve been pumping.
Not hard to understand why PHE (just as Ferguson) continues to be quoted and believed…just have to go back to who it is that controls the mainstream media…just about EVERYwhere.
The Ministry of Truth is interested ONLY in the “Truth,” but one must remember their teaching — Truth is a Lie, and Lies are the Truth. Newspeak prevails. Don’t forget that War is Peace, Freedom is Slavery, Ignorance is Strength.
What the data say about asymptomatic COVID infectionshttps://www.nature.com/articles/d41586-020-03141-3
and
Asymptomatic transmission of covid-19https://www.bmj.com/content/371/bmj.m4851
have put the asymptomatic spread of COVID at around 17%, although much, much higher figures were used to justify the lockdowns. Now that COVID is not being used as the primary weapon to defeat the President, it is possible that similar, more truthful studies will emerge.
Imagine that!
The British House of Lords official policy is that President Trump must not be allowed a second term. It sure looks like the revision from 81% to 17% is timed with the vote fraud.
British voters do indeed pay a heavy price for geopolitics, I’ll say!
One price that’s being paid is the remorseless pumping of extraordinary quantities of self evident, self serving virus bilge through the UK MSM – relying in large part on people’s apparent inability to recall what happened last week / month. Over 200 UK local newspapers are operated by Gannet Corp of McLean, Virginia – on the evidence, a short leash in being used.
Way to bury the lede. This is NOT a “new” variant, but the THIRD variant. What happened to the other two? Not deadly enough to create Panic Porn?
Not “likely to be resistant to existing vaccines”? How about we FIRST prove the vaccines to be “safe and effective” against the variant they were designed to immunize against.
Bottom line: The experts still don’t know sh.. about the disease and are just farting guesses out their a…. on how to combat it while ignoring safe and effective treatments for it.
But heck, I’m either preaching to the choir or my words are falling on deaf ears.
But at least we have masks.
In the UK intensive care wards are again being overwhelmed with covid19 patients as in the spring.
Therefore stronger quarantine and lockdown measures are needed, it doesn’t matter what the R numbers are or the details of the virus strains.
Most infections occur in homes. Source: https://www.thedailybeast.com/italy-did-everything-right-to-stop-a-second-wave-of-the-coronavirus-so-what-went-wrong Therefore, lockdowns actually increase the spread of the disease because people spend less time in social settings where the spread is much less.
I wonder if masks are too. Ever since “mask up” cases have exploded.
The kind of masks most people are wearing are just more of the same fear scam. A study in Nature a few weeks ago showed that virus particles that you cough into the mask would collect on the cloth fibers and be expelled on the fibers by normal breathing. So they probably greatly increase the spread from expelled droplets.
And that’s before you consider that the masks result in people touching their faces and eyes and therefore infecting themselves when they wouldn’t have done without the mask.
Remember how, nine months ago, the ‘experts’ who are now telling us we have to wear muzzles were telling us that masks would either be pointless or actually make things worse?
Oh Phil,
The intensive care wards are being “overwhelmed” by the usual winter flu victims Would it interest you to know that the actual number of fit and healthy English under the age of 60 who have succumbed to Wuhan flu is actually 388 as last week. A presenter on a London radio station who stayed that official figure was almost hanged drawn and quartered.
Expect LBC to go down the memory hole unless they follow the party line.
“Therefore stronger quarantine and lockdown measures are needed”
What’s really needed is an official policy of Early Treatment of the Wuhan virus as soon as a person tests positive, and this will lower the number of people having to go into the hospital.
Tom and Phil, I am not certain hospitals are ” overwhelmed” more then usual in a strong flu year.
Link shows December US hospital ICU occupancy at 72 percent. ( A bit higher now.)
https://www.statista.com/chart/23746/icu-bed-occupancy-rates-in-us-hospital-areas/
While this study…
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3840149/
shows ICU occupancy typically ranges from 52% to 82% and the percentage of patients on respiratory assistance ranges from 22 to 39 percent.
Yes, some hospitals are overwhelmed, yet this often happens. I am not yet convinced that our hospitals are being overrun.
California is overwhelmed, at least the southern part. I heard they are requesting that the hospital ship Trump sent them, be sent back.
They ought to be requesting therapeutics, to be used in Early Treatment, too.
It’s a crime that Early Treatment with therapeutics has been demonized to the extent it has. It’s inhumane.
Like this ?
https://t.co/dBnf9WqHKq
Considering that SARS-CoV-2 is likely endemic to some Chinese bat population and their ACE2 conformation, it has long been predicted that SARS-Cov-2 will stably adopt more human-specific mutations. Given that enough circulation in the human population has now occurred, not surprising that the virus has been selected to a more optimal Spike-humanACE2 binding. and thus we’re now seeing the emergence in population of mutations, like the N501Y variant, with the increased likelihood of transmission as an expected result.
SARS-CoV-2 will be endemic to the human population, just as the other 4 pre-existing Corona viruses are already. The vaccine (especially the mRNA versions), being antibody centric, will have limited period of efficacy in any peson who gets it, a few years at best. The Astra-Zeneca vaccine in the UK, likely has the best chance of eliciting a longer-term immunity to recipients.
The chance of a “Thanksgiving surge” here in the US (after our Nov 26th holiday) finally provided me the impetus to create a web page showing the exponential growth and decay rate of Covid-19 cases. The data involves taking the derivative of the new cases, and derivatives are noisy enough so it is hard to pick out the signal. Another complication is that several states didn’t publish data for either Thanksgiving or Christmas, and in some data sources that leads to a dip through the holiday and a spike after it.
So, I’m not convinced my graphs are good for that. I’ll add GBR graphs today in case that picks up on a increase (new strain) or decrease (new restrictions) in their case rate.
Check out http://wermenh.com/covid.html if you’re interested.
The growth/decay rates are plotted as a percentage change per day with a 13 day weighted average with background colors for the weeks per doubling (or halving) of the new case count.
Yeah daily doesn’t work too well because holidays affected both testing and reporting numbers. Some states don’t report daily in the first place (e.g., Rhode Island).
Nice work Ric, I live in VT and appreciate looking at the data (such as it is…).
What do you think of the disappearance of the regular flu season, isn’t that a bit strange, reporting wise? What does it say about the validity of the testing protocols?
While I’m familiar with the arguments that says wearing masks and telling people to stay home are ineffective, I think there’s a fair amount of empirical evidence that says they actually do work. As such, I suspect that’s helping to keep seasonal flu at bay. It’s not completely gone, per the CDC, it just not following the usual pattern.
There seem to be some people claiming that what we’re seeing now is influenza, but the symptoms are quite different once up to speed (e.g. flu doesn’t disrupt your sense of smell, trigger blood clots, or get people on ventilators for weeks).
I don’t know if things have changed this year, but flu cases haven’t been tracked closely or always reported. This would be a good flu season to do so. The influenza tests at doctors’ offices are antigen tests, good enough for most purposes, but not perfect. Possibly okay for typical uses, maybe not for research studies.
I don’t have too much trouble with the PCR tests, even when a lot of cycles are used. If they were problematic, they probably couldn’t be used to mark when a patient is cured.
Slightly off topic:
Gov. Tony Evers signed Executive Order #101 ordering the flags of the United States and the state of Wisconsin to be flown at half-staff in honor of Fire Chief Donald Edwin Kittelson of the Clayton Volunteer Fire Department who passed away Dec. 17, 2020, after contracting COVID-19 in the line of duty.
Best Case Scenario
A highly infective variant with much lower severity* than SARS-CoV-2 original and confers immunity to SARS-CoV-2 original. A “natural” vaccination.
___
*like a common cold.
The main difference of the new virus is that it does infect the kids.
Whether the (western) vaccines work against it is still to be shown.
The russian vaccine does.
got a link for that claim?
seems like alot of WAG’s involved in all of these studies based on an assumption that they know exactly how effective every mitigation measure is (which they don’t) … lots of hand waving by “experts” who can’t even say if asymptomatics are or aren’t infectious (they aren’t but the experts are terrified to admit it)
See above Nature and BMJ reports – geopolitics makes utter fools of them all.
Is there a single instance of where the SARS-CoV-2 virus has been isolated into a gold standard? Any at all? And we are here discussing strains?
As the original Covid-19 was never isolated, cultured, and shown to cause Covid disease, how in heck can they detect variants? Likely variants are simply their recognition of the family of coronaviruses out there. You HAVE to have a Gold Standard virus, if you want to pretend there are variants out there.
Also, we know in virology that more infectious also means less virulent, which is a good thing. As the crappy nonspecific PCR test detects even rhinovirus and general coronaviruses, pretending there is a variant is fraud, as they really do not know anything. They are saying the virus has mutated and is now more like a 24 or 48-hour flu. No, the original virus is gone with the seasons and the new flu season is here. Duh.
I’m at a loss to understand what you the anonymous astonerii are griping about. It’s obviously been adequately isolated to decode its genome many, many times. Check out https://nextstrain.org/ncov/global to see their tree’s root and many variants.
The Phase III trials show that parts of the genome trigger an immune response that prevents people from coming down with Covid-19. How do you propose people test to see if an isolate causes Covid-19, good luck getting that past the ethics boards. Perhaps you two could volunteer.
“Also, we know in virology that more infectious also means less virulent.” I’m not a virologist, please provide a reference to that. It would seem to me that increasing the binding strength of the spike protein should not have much impact on the course of the infection.
If you want to get a genome you have to have a 100% pure sample. Frequently called a gold standard. What they are doing is the equivalent of taking a frog, a cat, a lizard, and fish and blending them all together into a puree and then telling you that they can then pull out of that the DNA of a unicorn.
Charles, I am just curious where you get your information. The original virus has been isolated and decoded, along with countless variants that are constantly occurring.
Also, it is true that typically as a disease gets more infectious it becomes less virulent. But that isn’t a rule. There are examples where an infectious disease becomes more virulent and more infectious. The jump to syphilis appears to be one such example. Also, there are about a dozen different PCR tests. Are you really sure you want to suggest that they all detect rhino viruses and general coronaviruses? In other words, none of them are very specific.
I like Nics’ analysis but in a few days time we will know.
Not long ago this website published and supported analysis that said herd immunity was imminent.
That proved to be wrong.
Yet daily cases now are about 600 percent above the first wave, and daily deaths have only trended up 10 to 80 percent.
Some have suggested that the worst areas of the first wave are not experienceing the 2nd wave anything like areas of lower exposure to the first wave. ( I have not seen this verified)
Treatments have not improved that much, except where HCQ, vitamin D, and Ivermectin are used correctly, where the death rate drops to no more then a regular flu.
Nic’s work as usual above my head, but perhaps he’d appear out of the ether to answer a few questions for a layman. His conclusion echoes findings that the “new” virus appears to be notable for “higher viral growth rates” and “viral loads”, with likely faster transmissibility. I’m curious why these factors don’t translate into more “virulent” strain.
Regarding the “new” strain’s newness: I see Colorado, as of last night, now can claim the honor of being the state where the “new” virus has first reared its spiky head. To me this raises questions of verifiability. The “new” B.1.1.7 was first identified in Britain in later September, (before UK travel bans were announced?). In terms of virus generations, how is that considered “new” in late December? (and) Wouldn’t it be logical to assume that a strain that popped up in late September in Britain is well and truly mixed into the U.S. cocktail of viruses already?
To this point… I heard Fauci the other day addressing a reporter’s question, to wit: we aren’t testing for different strains of the virus in the U.S. the way they are in the UK. In heaven’s name, WHY? Isn’t this the basis of the vaccine’s efficacy? Aren’t we interested in the virus’s mutations? One of several articles on this conundrum:
What scientists still want to know about the new … https://external-content.duckduckgo.com/ip3/www.nbcnews.com.ico https://www.nbcnews.com/health/health-news/what-scientists-still-want-know-about-new-coronavirus-variant-u-n1252122
The U.K. has a robust system in place to monitor for genetic changes in the virus; in the U.S., however, just 0.3 percent of infections have been genetically sequenced, according to the CDC. That…
“Wouldn’t it be logical to assume that a strain that popped up in late September in Britain is well and truly mixed into the U.S. cocktail of viruses already?”
Yes, it would. B.1.1.7 is called a new variant because it has only recently been identified. There are thousands of different mutations, and too many variants to worry about almost all individual ones. Until the fact that a new variant has arisen in enough cases and is behaving differently (which may be a few months after it first arose), it usually doesn’t attract attention.
A confounding factor is that people who get symptoms do not get a test because they do not want all the hassle and lack of freedom that comes with a positive test result.
It is not the restrictive regulations that have an effect on transmission, it is how people actually behave, and from surveys only about 10% of people comply.
I read Nic’s last analysis in October that sort of suggested herd immunity for London etc might be imminent. As a prediction that hasn’t gone too well. London was badly affected in first wave, serum tests indicated 17%+ of population of London had been infected. London is now badly affected again, so where was the predicted herd immunity?.
The 2nd wave virus has invaded my own family and close contacts, whereas the 1st wave only a few isolated contacts got it, my cousin is dead before her time and at this time 6 family members are ill. They all live in relatively low incidence areas.
History tells us that the 2nd wave of an epidemic always kills more than the first.
John, would you link to any source for this assertion? “History tells us that the 2nd wave of an epidemic always kills more than the first.”
Apparently this was true for the 1918 pandemic. But it’s a strange phenomenon that interests me. There appear to have been three major humps in the graph of “Spanish flu”, all of them occurring over the span of less than 2 years.
John Barry, in his Great Influenza, traces its first appearance in Kansas, June – July of 1918, and the following infection of US army cantonments, it was like any other flu – dangerous, but not as deadly as the second wave (of its presumed mutation) which occurred over months of Oct, Nov, Dec and comprised the bulk of the deaths; the third wave, in the spring of 1919, killed nearly half as many as the second.
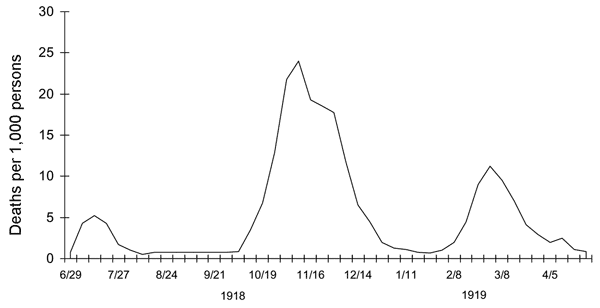
Barry explains the etiology this way: a virus may become more virulent after it undergoes a certain number of “passages” between hosts. During these passages, it learns to be better at evading the existing immunity levels in the host population. The process continues until the virus becomes too deadly for the existing population, whereupon it attenuates to survive. When that version of the virus becomes predominant, it may be more efficient at transmission from one individual host to the next, it has become less virulent. Thus the third wave can in general represent the “echo” of the former ones as the virus is on its way out.
If this jibes with what you’ve read, perhaps you’d comment. I’m no expert. Just interested.
BTW, he also notes that the epidemic waves tend to fall off abruptly, not slowly. In looking at the high-transmissibility + low fatality factor, as well as the current 7-day average of daily new cases… might give us some reason to hope this thing is soon to taper off. (?)
I think the “fall off abruptly” phase reflects herd immunity. In this pandemic we’ve likely done a lot more to slow the spread because the promise of making a vaccine makes that worthwhile. In other pandemics, People were pretty much going to get infected and it didn’t matter much if they got it sooner or later. So we haven’t achieved herd immunity, things are spreading for likely several reasons, and I’d like to get vaccinated sooner than later.
Despite the detailed analysis presented here, I somehow strongly doubt of the existence of this new variant.
Here is a simple syllogism:
(A) The flu viruses (5 strains of influenza) have virtually stopped circulating worldwide (per WHO website) and in the US (per CDC) since the early global shutdown of international flights and ban of social gatherings in mid-April. (*)
(B) The coronaviruses (old and new variant) spread similarly as flu viruses.
(A+B) Thus coronaviruses must have also stopped spreading since mid-April.
How the coronaviruses could continue to circulate and new variants to start popping out from nowhere while all strain of flu viruses have gone off the radar?
In my opinion, the only logical explanation is that the COVID-19+ qPCR tests are 100% false positives (old and new variant). It looks like if the imaginary whooping cough epidemic at the Dartmouth-Hitchcock Medical Center Hospital, Lebanon NH in 2006 is now repeating itself but on a worldwide scale. As in the NH hospital, where the whole medical staff have been tricked in believing the reality of the whooping cough using qPCR, so is the entire world right now of this COVID-19 pandemic. The title of the NYT article reporting this story: “Faith in Quick Test Leads to Epidemic That Wasn’t”, would nicely summarizes the 2020 COVID hysteria.
As for the “COVID deaths”, it has been repeatedly pointed out on this website (one of the best covering the COVID topic) that most are unrelated to COVID. This is corroborated by the fact that in several countries the average age of COVID deaths exceeds life expectancy.
I guess the silver lining of this overplayed pandemic is that the flu and other transmissible diseases, like GI tract infections (no more norovirus outbreaks in US per CDC website), may have been eliminated for some time.
(*) During the holidays, the circulating flu cases should be “skyrocketting”, there are barely nothing currently. This is not a consequence of receiving less samples for analysis, they are analyzing as many as usual. The % of positivity is simply extremely low (perhaps just reading noise). Note that they most likely use a hemagglutination assay not qPCR.
Wisebear,
Your analogy to the Whooping Cough panic is apt. But I think the NYTimes article makes the point rather glibly that the original diagnostic error, attributed to an unnamed “quick test”, was only corrected later by a “definitive test”, a laboratory culture of the bacteria. We’re facing the same question now about “quick” vs. “gold standard” testing. In my mind the article raises important questions about how quick tests are administered and interpreted.
There appear to be many quick tests for Covid on the market now, some of which would have us quarantining all the Kiwis (the fruit – not the islanders). At the same time, the gold standart PCR seems to deliver positives for anybody who’s ever had Covid even if they didn’t know it and recovered from it 8 month ago. This isn’t exactly a “false positive”, but it’s a wild goose chase, since all that’s being read are inert and non-transmissible fragments of the virus’s DNA. False positives from either test would be enough to make a person skeptical about the disease itself. But you shouldn’t be.
At some point, some medical student is going to do a retrospective analysis of your question, but phrased more like, “How many Covid patients were actually just Flu?” It’s a shame that such a study isn’t being conducted currently (that I know of) to gain a basic understanding of who is being admitted to hospitals and for what? The test should ideally follow pulmonary patients over two or three months for flu, for Covid-19, and for bacterial and viral pneumonia. Such test would be run: a) on admission b) weekly until discharage c) for, say, three months after discharge. Subjects could be ruled out for co-morbidities. The tests should be able to distinguish clearly between all three diseases so that a nosocomial appearance of one of the other two diseases would be diagnosed promptly. If a person had only flu upon admission but no positives for Covid, both quick and PCR tests would show “negative”, validating each other. There are many tests for flu. Such analysis could tell us a lot about our hospital systems, including the percentages of infections acquired in the hospital. And complementary testing methods would reinforce each other. If a person showed only Covid but no flu at admission… well, you get the point. We’d know for sure what we are dealing with.
This would take some man-power away from harried hospital workers perhaps, but it would be worth it. And if it’s being done already, I’d sure like to see it.
There is some data in the UK for Healthcare Acquired Infections of COVID-19. The two articles following use different definitions, and recognise that they are likely to miss many cases, but are consistent with a significant proportion of COVID-19 hospitalisations in the NHS as having been infected in hospital.
https://www.cebm.net/covid-19/the-ongoing-problem-of-hospital-acquired-infections-across-the-uk/
https://www.hsj.co.uk/patient-safety/covid-infections-caught-in-hospital-rise-by-a-third-in-one-week/7029211.article
While the rates vary over time and between hospitals, they do seem to rise above a quarter and even a third of hospitalised cases in several instances.
Paul C Thank you. Just saw this. From Google searches and these two articles it appears that the rates of nosocomial infections are on the rise everywhere. I don’t know how it could be otherwise unless patients are assigned “dedicated” staff to tend to one patient only.
Still have questions re types of testing being used and the virtual disappearance of flu.
Sidebar says, ” As for the “COVID deaths”, it has been repeatedly pointed out on this website (one of the best covering the COVID topic) that most are unrelated to COVID. ”
Sorry, yet that is very much mistated. What is asserted is that most Cov19 deaths have contributing factors. Also, Cov19 is known to create and exasperate contributing factors, like pneumonia, heart issues, etc…
That is certainly not the same as saying the deaths are not related to Cov19.
David A
I understand that in the US hospitals received more money from Federal (?) funds if a person died from covid 19. Here in the UK the NHS also has a special fund for covid deaths and I have been given to understand that cash strapped hospitals are “using it”.
FWIW, NH recorded its first flu death this week. I haven’t looked into details, but will look at the next weekly CDC influenza report with a little more interest.
I know correlation is not causation but the correlation of new variant dominance and high R values in the SE, E and London areas looks pretty persuasive to me as does the London R values in November when under unchanged lockdown conditions R went from around 0.8 to around 1.1.
And yet our London-centric government avoided putting London into Tier 3 until a couple of weeks later whilst most of Northern England had no respite between the national London lockdown, and the virtual lockdown of Tier 3 in spite of having lower infection rates. Not that lockdowns do much to inhibit the spread of a seasonal respiratory virus. They seem to be positioning the rollout of vaccines to take credit for the seasonal reduction in infections in spring.
Figure 2 in the 2nd PHE Technical Briefing (https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/948152/Technical_Briefing_VOC202012-2_Briefing_2_FINAL.pdf) has data right up to week 52 (ending 27 Dec). It shows that in week 52 the proportion of new cases represented by S gene dropout (almost 100% of which represent the new B.1.1.7 variant) fell from 58.1% in the previous week to 56.2%. This suggests that the new variants may no longer be growing relative to existing variants, although it is only one week’s data.
CDC- Deaths from choking on food higher than dying from Covid for the under 50s. When is this Covid nonsense going to end.
Lessee, https://www.statista.com/statistics/527321/deaths-due-to-choking-in-the-us/ shows about 5,000 choking deaths per year for all age groups.
https://covid.cdc.gov/covid-data-tracker/index.html#demographics says 6,800 deaths so far in the age 40-49 age group.
Please post data sources when you make claims that sound bogus.
I turned 50 decades ago (okay, two decades ago). Why don’t I count? Get off my lawn.
50-64: 34,000
75-84: 65,000
85+: 78,000
This Covid nonsense will end when nearly everyone gets vaccinated.
Who cares about the elderly anyway? The only people who should be taken care of are the ones in the working ages-18/65-, in the UK, for instance, let us start from the top… oh my God!
Your doctor has clearly been trying to kill you all along. Or is suicidal. Or both. Other than that, situation normal. Everything’s fine. We’re all fine here. How are you?
I know that because the director of the CDC, Dr. Robert Redfield, told me so this week when he said masks are even better than vaccines. Because I simply don’t remember a flu season when I saw doctors regularly wearing masks as a routine preventive measure. Not at casual doctor’s office visits (where the flu is more dangerous to children than COVID). Not at parent-teacher conferences or youth sports games (my God, why do doctors hate children so much?). Not at the golf course (foursome or death wish? You decide).
What utter psychopaths. They spend all day telling you to lose weight, eat better, and stop smoking, only to go out in the world and breathe hellfire into the universe.
Clearly, doctors should never, ever be able to take their masks off from now on, especially since we have learned so much cool woke science since March. I mean, Karen has reliably told me that I have to wear a mask so the virus doesn’t jump the six degrees of separation between me and Kevin Bacon. Meanwhile, should I spend time exercising outside in the vitamin D-packed sunshine, I risk infecting my grandma, who is riddled with co-morbidities.
And I think I can speak for all of us when I say thank Allah for Karen’s nagging persistence on that front. Such a blessing. Redfield is only now catching up with her wisdom.
So what the hell, doc?! You are meeting with my co-morbid grandma and everyone else’s co-morbid grandma in your practice every single day, as the flu just plays Russian roulette with the maskless lot of you? Have you no decency? Have you no shame? It’s just if you were conducting open heart surgery without a mask on, coughing your foulness into a vulnerable chest cavity. May as well at this point. Clearly nothing matters to you monsters.
In a way, we are all chest cavities now. So the doctor without a 24/7 mask on, no matter where he goes and no matter what he does, is basically no better than a jihadist. Are we all clear on this? I mean, it’s just science. And the science, as always, is settled. Until it’s a very fluid situation, of course.
Hard to believe a medical establishment that was finally getting it right on the fact that a man can have a uterus has been so in the dark about the fact we beat the Black Plague with masks.
Perhaps all doctors should now live together in communes if they can’t otherwise embrace the seriousness of their craft. What a failure the Hippocratic Oath has been, not to mandate that doctors wear masks at all times all these years. Clearly, lives of chastity, poverty, and total obedience may be in order to bring greater focus and irrational obsession to keeping me safe from everything I imagine that isn’t remotely real.
The doctor has spoken. All hail the mask. Now pass the Kool-Aid and give me something cool like a comet to worship.
This is an excerpt from my latest missive sent last week to our lockdown-and-vaccine-addicted Alberta government.
Apologies, but I’ll have to read Nic Lewis’s post later. Hope we agree.
Subject: END HARMFUL LOCKDOWNS NOW – 31 – COVID-19 mRNA VACCINES (PFIZER AND MODERNA) ARE HIGH-RISK – UNKNOWN FUTURE SIDE-EFFECTS
SUMMARY AND RECOMMENDATIONS RE COVID-19 – SEE SCORECARD BELOW
There is no real Covid-19 pandemic. Covid-19 was only dangerous to the very elderly and infirm, and is similar in average mortality to other seasonal flu’s of recent decades.
The Covid-19 PCR test is not fit-for-purpose and provides many false positives. Routine testing of asymptomatic people is a waste of resources and drives erroneous policies including lockdowns.
The Covid-19 lockdowns were never effective or justified. Harm done by the lockdowns exceeds by 10 to 100 times the harm from Covid-19. End all lockdowns now and do not lockdown again.
Simple, inexpensive treatments are known to save lives – Vitamin D, Ivermectin etc. Why are these treatments not being widely recommended and implemented by Alberta authorities?
The increase in deaths of the elderly in Winter is a well-established seasonal phenomenon. “Excess Winter Deaths” in the four Winter months routinely average about 100,000 per year in the USA and about 10,000 per year in Canada, as described on our 2015 summary of Excess Winter Mortality that includes the landmark Lancet study.
COLD WEATHER KILLS 20 TIMES AS MANY PEOPLE AS HOT WEATHER
https://friendsofsciencecalgary.files.wordpress.com/2015/09/cold-weather-kills-macrae-daleo-4sept2015-final.pdf
The Covid-19 vaccine developments were rushed and are not proven safe or effective and should NOT be taken, especially by the low-risk population – those under-65 or recovered from Covid-19. The two experimental Covid-19 vaccines that contain mRNA (Pfizer and Moderna) are especially risky – due to unknown future side-effects, the risk-to-reward is far too high for the low-risk group. The Oxford-AstraZeneca vaccine does not contain mRNA and is expected to be more safe.
_________________________________