Guest post by Gregory J. Rummo.

It was one week after the terrorist attacks on 9/11 when envelopes containing a white powder began showing up at random locations in four states; among them, a newspaper office in Florida, the Washington D.C. office of then-Senate Majority Leader Tom Daschle, NBC News and the New York Post. The white powder turned out to be anthrax spores, engineered to be readily dispersed and inhaled – a potentially deadly bioterrorism weapon.
Anthrax infections are treated with antibiotics. There are two that are most effective; ciprofloxacin and doxycycline. At that time, I was the CEO of a small pharmaceutical company that represented foreign API manufacturers in the US. We had a large, domestic customer base to which we marketed dozens of anti-infective agents, including antibiotics. Doxycycline was one of them.
We had been working with Zenith Laboratories, a generic pharmaceutical manufacturer in South Florida (currently a part of Teva Pharmaceuticals) to approve our doxycycline for use in its formulations. Normally, the turn-around time for the FDA to approve a drug, even a generic copy of an existing drug (which doxycycline was), is well over a year and often two.
But this was different. The US was facing a crisis in the form of a potential bioterrorism attack. The federal government’s response to anthrax quickly became a national emergency. Zenith Laboratories, along with other manufacturers, was awarded a contract to supply tablets and capsules to the Department of Defense’s Strategic National Stockpile. In less than one month, the Food and Drug Administration granted an emergency use authorization and overnight, we became approved suppliers of doxycycline.
Fortunately, anthrax never became the bioterrorism threat many had feared. Five people died as a result of coming into contact with envelopes contaminated with the spores that had been delivered through the postal system.
Our government’s coordinated response in 2001 to apply pressure to drug manufacturers and its own Food and Drug Administration to expedite approval of a life-saving treatment for a bioterrorism weapon bears an eerie similarity to the national health crisis in which we find ourselves.
Any drug, be it an antibiotic, a monoclonal antibody cocktail or a vaccine goes through a rigorous, scientific process long before ever falling into the hands of government regulators, let alone politicians. Drug development begins with a conceptual design model followed by research, engineering, small scale manufacturing and several phases of testing; usually first in animals and then humans. Failures are common along every step of this process. By some estimates over 90 percent of drugs never make it to market. Safety and efficacy must be demonstrated by the manufacturer before the FDA will approve any drug’s use in the general public.
What is currently known about the two leading candidates’ mRNA vaccines—Pfizer and Moderna Inc.—is that they are showing safety and efficacy in late stages of phase 3 clinical trials.
Participants like me have received the first and second booster injections and have provided blood samples to researchers. It is no stretch to believe we will have coronavirus vaccines approved under the FDA’s emergency authorization use as early as next month for distribution to, at the very least, healthcare workers and those most at risk of severe morbidity.
This is not politics but, in fact, the result of science – lots of science – and shame on those politicians who continue to make this an issue of anything but.
Gregory J. Rummo is a Lecturer of Chemistry at Palm Beach Atlantic University and a Contributing Writer for The Cornwall Alliance for the Stewardship of Creation. He is the former CEO of New Chemic US Inc and patient 001 in Moderna’s mRNA-1273 Phase 3 trial currently being conducted at the Palm Beach Research Center in West Palm Beach, FL. The views expressed in his columns are his own.
I’ll believe it when I see it and it’s effectiveness though proper antibody testing.
Funny how the antibody testing hasn’t been congruent with vaccine development or even outpaced it and has all but disappeared from the headlines.
It is a double blind study so there are no official reports about neutralizing antibodies. But informal comments from the healthcare professionals working at this clinical trial site are that it is producing very positive results. Let’s pray!
Agree (except “let’s pray”)
Positive antibodies do not mean very much:
– they may not confer 50%+ immunity [50% being the (low) bar currently set by FDA]
– there still may be strong side-effects
– they may not work for various demographics (e.g., the 65% of Americans who are overweight or obese, the 18% of American adults who are smokers)
– any vaccine will not be widely available for a year
why do I immerse myself in this minutia? I’m a subject of the Moderna vaccine trial.
BTW, why is this a topic for a Climate site?
About Watts Up With That? News and commentary on puzzling things in life, nature, science, weather, climate change, technology, and recent news by Anthony Watts
This science news site feature original content from myself as well as several contributors:
right in the about section
AFAICR, that anthrax sample was traced back to being a military grade strain of US origin, once the DNA markers were fully analysed.
A bit like explosives, there are traces deliberately built into these things to allow detection and identification of sources.
Hopefully one day we will be able to work out exactly what lab produced the original strain of SARS-COV2. Let’s pray!
One thing is for sure, courtesy of the ‘immaculate furin linkage’ in SARS-CoV-2, it was definitely ‘made in Wuhan’….
seemsto me these are the ONLY vaccines theyve even bothered to do the double blind on?
bit hard to do when the trial object has NO other actual similars to BE used for comparison either?
well standard h1n1 etc but theyre NOT in the same field are they
Since they do NOT have the virus in pure culture to work from, how could they possibly make a vaccine? That’s a nonstarter.
Then, the PCR test is NOT based on one virus but also responds to covis, the common cold, and even some human DNA.
The antibody test, again because they have no pure culture, is NOT specific for anything.
It’s all a joke. Sure a vaccine might make one produce antibodies, but, as they do not have the virus in question, there is zero proof it protects one from a virus they do not have or actually identified.
Also, as the flu season salad of viruses has been around the world and a new flu season beginning, where are they going to get people who are not naturally immune, not specficially immune to whatever virus, and ready to be infected? Again, this is a cosmic joke on the world.
Charles
I have posted here before and no one is following up. So, I’ll try again albeit with a little more info. Here are CDC websites that back up exactly what you are writing (though the website is for the FDA, it is a CDC report from July 12/13, 2020):
https://www.fda.gov/media/134922/download
especially page 38 of report (page 39 on Adobe Acrobat):
“Detection of viral RNA may not indicate the presence of infectious virus or that 2019-nCoV is the causative agent for clinical symptoms. ”
AND:
page 39 of same report (page 40 on Acrobat reader) : “Since no quantified virus isolates of the 2019-nCoV are currently available…”
Lastly, you cannot have a vaccine if it’s based on the following FAKE SCIENCE (also from the CDC, yes, wwwnc.cdc.gov is legitimate and is a backdoor to some of their reports which are otherwise difficult to find):
SEE:
a. https://wwwnc.cdc.gov/eid/article/26/6/20-0516_article
b. https://breakawayindividual.com/2020/10/20/dr-tom-cowan-explores-the-covid-virus-invented-out-of-sheer-nonsense-covid-19-coronavirus-sars-cov-2/
c. https://drtomcowan.com/only-poisoned-monkey-kidney-cells-grew-the-virus/
The above show that the “genome” of “covid-19” was come about by identifying 37 pairs of approximately 30,000 pairs of genes on the RNA that was taken. The rest was “filled-in” by software giving virologists several choices of possible genome to choose from and these virologists (20, I think) agreed by consensus (“vote” — sounds like global warming rubbish methods) which one they “thought” would be right. This is farcical and is what the false rt-PCR “test” is based upon.
2. When this bit of tissue was taken and analyzed and grown in lab in different cell tissues it resulted in NO INFECTION of human tissues and only caused a problem with monkey kidney cells which had also been injected with two toxins known to cause damage to kidney cells.
By the way, 37 of 30,000 pairs in the genome amount to just over 0.1% of the genome — humans are about 96% related through the genome to rats (why we test them for medicine, etc…) and about 92% to the chimpanzee. How does 0.1% possibly right come up with 100% positive? Because of the possibility within each pair of difference in order within the pair, this means 0.1% is much,much too generous for showing similarity genetically.
Without isolation, without proof of a genome, without proof that what cannot be isolated or proved to exist actually causes infection is beyond science and is more like hallucination. How can one make a vaccine on this? How can one test a vaccine such as this. This smells of Solyndra x10.
Give me a break!
AK in VT
Great summary & well researched -thx ! The short life of antibodies being generated in the human body is also a major challenge for any vaccine….good luck developing one to last over 12 months…
https://www.sciencedirect.com/science/article/abs/pii/S0140673620302518
Charles and others
I wish to post this ink which will logically explain better what I have posted already. This is another reason why a vaccine is pointless and this whole “covid-19” thing is sham “science.”
Try this for more detail and links (much will be repetitive to what I have already written, but there’s much, much more):
https://blog.nomorefakenews.com/2020/10/22/the-virus-that-isnt-there-genetic-sequencing-and-the-magic-trick/
Yes, Jon Rappaport does not believe in vaccines (or, at least, the majority of them), but that does not mean what he has to write and his CITATIONS are not worthy of looking into — after all, is this not what WattsUpWithThat.com is all about?
Regards
AK in VT
Interesting blog thx. Seems to live up to its name….No more fake news.
Which is why they’re doing trials. Maybe you missed that part.
dont confuse them with facts
He seems to be questioning the validity of the trails when there is no available isolation of the virus to base any trial on, maybe you missed that part.
Contrary to reports of isolation in the media, subsiquent FOI requests to confirm virus isolation reveal no such isolation has been carried out…
https://www.fluoridefreepeel.ca/university-of-toronto-sunnybrook-hsc-have-no-record-of-covid-19-virus-isolation/
I hope you don’t get confused by those facts
Vaccine trials to prove what? That there is success in stopping a virus (which has not been proven to exist) from infecting someone. Fantastic! What a no brainer to pillage the US Treasury (aka: you, the taxpayer):
1. say there’s a virus
2. get the media behind you (they need ad money because their ratings are way down and everyone will tune in to see what’s happening around the world)
3. get the CDC involved (the CDC exists pretty much soley for the medical industry to make money)
4. claim the virus has more symptoms than anything else we’ve ever discovered (last I checked it was 13 major symptoms and that was back in April! — wonder how mah it’s up to now), that way you can claim all these deaths are due to “covid-19”
5. get everyone so disturbed they practically cry out for anything to help them
6. VACCINE TO THE RESCUE!!!
7. Vaccine cannot fail because:
a. the taxpayer is going to pay for all the research and most of the distribution
b. if the vaccine makes people ill, we can say it was the virus having mutated
c. if after wide distribution of the vaccine the “case numbers” drop, it must be the vaccine!!!
despite the fact that the PCR “test” can be tweaked by lower the amplification any time the labs want to that no “infection” shows up — just like they have raised the amplification much higher than has ever been done before (which, pf course, makes it seem like “everyone” is infected)
How can the efficacy of a vaccine produced to combat a virus that doesn’t exist be calculated — it will always be a winner.
Major scam — just follow the links above on my earlier posts. These links are legitimate CDC and FDA links which pretty much explain to the logical mind that this is all a hoax.
Regards
AK in VT
I believe it, in the military we have already been given a heads up, to be ready to assist in delivery and to receive vaccine for ourselves. If we are receiving white letters from our top Generals, which by the way are released as (c) FOUO, mainly so it does not appear that the military is playing politics. It’s about optics as much as it is about the mission. The military does not want to get dragged into even by accident the political side of things. I can’t wait to receive mine so I no longer have to worry about getting close to people and especially get to finally lose the mask.
It’s real and it’s here. Once government gets out of the way Science can be good.
Global Warming is steeped in politics. Government needs to get out of the way and let the public innovate on their own, that is if Global Warming is as bad as they say it is. The problem is Government under liberal/socialist rule gets in the way because they want to do their own way, fund their own pet climate projects which unsurprisingly make them very wealthy. Global Warming has turned out to a cash cow for Dem’s/Green’s/ Socialists in America and around the world.
Good for Mr. Rummo, to put his “money” where his mouth is, in volunteering to be patient No. 1 in the Moderna Phase 3 coronavirus vaccine trial. This whole vaccine process was pushed along by project “Warp Speed”, as money was made available to advance in anticipation of positive results, instead of searching for new funding at every step. I note that the resurgence of cases in Europe is not matched by the same/similar resurgence in mortality (waiting for a delay between new cases and mortality data). This appears to suggest some mutation, leaving me to wonder how exactly the vaccines need to match the viral structure to be effective?
Possibly less mortality due to viral mutations, but more likely due to better treatments, less intubations, younger patients with less comorbidities and a population now educated about the importance of sufficient dietary zinc and vitamin D (among others).
yes to all that
but remember H1N1 managed to create a panic fast got some pharmas a massive profit from dodgy antivirals and by the following year its now a normal flu that makes a few crook/dead but majority no big deal
and the vax for that might be round 50% useful on average they admit now, it wasnt much chop last yr
That’s generous. The H1N1 vaccine killed and injured many poeple and is still being litigated in multiple damage cases. It is nowhere close to 50% effective. Many estimates put it at only 1-2% (the 50% figure comes from the fact that it reduced H1N1 from 2% of a study population to 1%. You can call that a 50% reduction but it still 1%). But maybe you’re speaking in the relative sense. Yeah the H1N1 vaccine is great compared to the swine flu vaccine administered in the 70s that paralyzed some 40000 people. You’re making me want to go get it right now! I’m already 99.9% immune to H1N1 and so is everyone else. Without even doing anything. But maybe that 1% protection it provides will get me to 100!
Most sensible oldies are being careful!
It is possibly the younger population mixing maskless at pubs music raves etc.
The bulk of the rise is due to these and the younger population do not die. see
https://coronadashboard.government.nl/landelijk/positief-geteste-mensen
new cases per age group (worst is 20 to 60 group)
No, the bulk of the rise in “cases” is the rise in testing. You have a PCR test that can be 85% to 100% false positives, but the authorities treat it as if its prefect and accurate. We have an epidemic of testing. If you test 1000 people and get 10% positive (100 people positive) and then the next day test 2000 people and get 10% (200 people positive), nothing is happening.
But, if you neglect to mention the total testing numbers and the proportion of negatives, you can shout, “OMG, the number of cases just went from 100 to 200 in one day! We must be stupid and lockdown again!” Complete propaganda by the authorities.
Exactly.
Cases, cases, cases, cases, cases, cases. Remember the key to ALL brainwashing techniques is REPETITION. And just because you know that does NOT mean you are not susceptible to it but at least you watch for it.
CaseDemic – https://www.youtube.com/watch?time_continue=483&v=FU3OibcindQ&feature=emb_logo
That’s because the control template is not COViD 19 but a human RNA seauence assembled by ‘consensus’ from known sequences from a library. Amazing that they match up to humans so well right!? The inventor of PCR said to never use PCR for this purpose. The test is baseless, factless and useless.
Almost ALL younger people think they are immortal and completely invincible! They don’t realize that they almost all have older relatives, and mixing in public exposes them to a high risk, too! Most of them think that only the older people catch Covid-19, but the news says different. There HAVE been young kids who catch it and some them, too , die from it, as well. Until we have RELIABLE, proven vaccines we can’t afford to let our guard down. Brazil just reported today that they now have over a million positive ‘cases’. It ain’t over til it’s OVER!
This is a disease with a 0.2% death rate. Where the average age of death is higher than the average life expectancy.
It only looked bad early in the year because many old, sick people were getting infected and then hospitals killed them by putting them on ventilators. In the general population it’s no worse than a flu.
So why can’t we afford to ‘let our guard down’? The response to the disease has turned out to be far, far worse than the disease itself.
Or do you plan to destroy what remains of the economy every flu season in future?
Ghalfrunt claims drinking bleach stops Covid so what does he know?
Here’s something else regarding “younger patients”
We are constantly being bombarded with information which states those younger patients who have contracted the virus, gotten ill then suffer permanent damage to their other organs – hearts lungs etc etc. shortens their life expectancy etc etc.
How do we know that those persons didn’t have underlying undetected issues or were predisposed to comorbidities and they, until now never gone through intense examinations or diagnosis to show up these health issues?
The western world is now seeing relatively younger people suffering from “old age” diseases caused by lifestyle and dietary problems. Young people in their twenties contracting type 2 diabetes and heart conditions normally reserved for the over fifties.
Obesity rates of western populations approaching 40% or more.
Are governments playing the nanny state game protecting lifestyle issue people?
At least one cardiologist I’m acquainted with has said. ” If we tested every 40 year old for heart problems, we might find ourselves with an epidemic.”.
https://www.thelastamericanvagabond.com/operation-warp-speed-is-using-a-cia-linked-contractor-to-keep-covid-19-vaccine-contracts-secret/
Look up;
gulf war syndrome. 20,000 solders dead.
13 dead from flu shot.
490,000 children have polio in India from vaccine.
No mutation. Virus is the same. Treatment has gotten better and the target population (oldies and comorbilities) knows the virus is after them and hide from the virus.
No model took good account of changes in behavior based on the info available. A person can avoid infection indefinitely through face masks and social distance. For all practical purposes he is immune.
Javier,
Even face mask supporters like the US CDC concede that face masks do not protect the wearer. As far as protecting others, the CDC guidelines state that face masks “may help reduce the spread” of viruses. Not exactly a strong statement; in fact, it’s not very reassuring at all.
The CDC is wrong on that one. Face masks have been used to protect the bearer from flu and other respiratory illnesses for decades in Japan and other Oriental countries. You only have to look at the flu data from this winter season in Australia. Face masks block droplets not aerosols but in doing so they reduce the danger of infection greatly. Together with hand hygiene and social distance, particularly the avoidance of being in closed spaces with people they make you almost immune to infection.
The problem is that the ones that are more likely to infect you are your family and friends. So the protection has to be against them also.
Increased hygiene and social distancing yes; masks not so much. I and many others have been driving to work since February now, and we go to the office only half a week. Our production has been spread on two shifts, to reduce the density of people in the workplace., and the two shifts are separated by one hour interval, when cleaning teams go through everything with disinfectant. Swipe cards are used to track people’s movements between different areas.
Yes. Masks protect so much.
Gandhi, M., Beyrer, C. and Goosby, E., 2020. Masks do more than protect others during COVID-19: Reducing the inoculum of SARS-CoV-2 to protect the wearer. Journal of general internal medicine, pp.1-4.
Chan, Jasper Fuk-Woo, et al. Surgical mask partition reduces the risk of non-contact transmission in a golden Syrian hamster model for Coronavirus Disease 2019 (COVID-19).” Clinical Infectious Diseases (2020).
Liang, Mingming, et al. Efficacy of face mask in preventing respiratory virus transmission: a systematic review and meta-analysis. Travel Medicine and Infectious Disease (2020): 101751.
MacIntyre, C. Raina, et al. “Face mask use and control of respiratory virus transmission in households.” Emerging infectious diseases 15.2 (2009): 233.
“Adherence to mask use was associated with a significantly reduced risk of influenza-like illness-associated infection.”
They have a long track record of usefulness against respiratory illnesses in Orient.
I know a lot of people don’t want to use masks. More power to them. The more of them becoming infected and developing resistance the less will remain susceptible to infect me.
Actually people in Japan wear masks against pollution not infection. Ironically Japanese have a genetic disposition to become infected with influenza.
and the kids who arent wearing masks
meanwhile we havent had the normal cold/flu season so our antibodies didnt get the usual kick/reminders
I dont think thats actually a good thing, weirdly Id actually like to catch a cold or normal flu, so things are “normal” antibody wise
Well, there’s one of the CDC mask guidelines that is studiously ignored by every single mask-Nazi in America. In fact, it’s number 4 on their list. To wit:
“Masks should NOT be worn by children under the age of 2 or anyone who has trouble breathing, is unconscious, incapacitated, or otherwise unable to remove the mask without assistance.”
My wife cannot breath while wearing a standard surgical mask. I used to be able to, for short periods. But I am very susceptible to bronchitis. Forced mask-wearing has turned my bronchitis into a severe chronic form. If I am forced to wear a mask for a long period of time (such as on an airline flight), it could kill me. I will no longer fly anywhere.
Yet the stats don’t support the idea that mask-wearing is effective. There have been some politically-hacked “studies” published recently to support the “mandates”, but the literature of the past three decades overwhelmingly show that masks are not effective (unless used by hospital-trained employees under very strict regimes, i.e. not by the general public).
Hi Michael: Please try taking NAC, about 1 gram per day and let me know in a week if your bronchitis is markedly better. I swear by it. It’s cheap and available at any vitamin store or most health stores. It well known to medical science. Anyone else want to chime in?
Mario, thanks very much for the tip. I just ordered some to try. Looks interesting!
Great… I started taking after learning about Covid 19. One of the prongs was to understand lung function. The NAC supports lungs almost magically.
There are other things that severely limit the RNA replication of the virus, and yet other things that boost our immune response.
I have bronchitis, including exercise induced, so this part of my research has led to my sinuses and lungs being better than I can remember in over 45 years.
Thank you. I have always believed actions should accompany words.
“some mutation, leaving me to wonder how exactly the vaccines need to match the viral structure to be effective”: In addition to Javier’s comment, mutations are by their nature almost all small. The likelihood of any given mutation (or the sum of several mutations) rendering a vaccine ineffective is near nil.
It does pay to keep in mind that an “effective” vaccine could be just 50% effective in clinical trials. And it might be better or worse in the general population. So even an effective vaccine isn’t perfect and might, for any given individual, be almost useless.
I believe that a vaccine may have a much higher efficacy among younger people with this rapidly diminishing with age. As this vaccine is not needed for probably some 99% of those under fifty and with limited effectiveness for those seventy and older – those most likely to die – I wonder if we have not squandered a huge amount of money that would have been better spent on finding better medications to treat the secondary infections?
All due respect to the author of this post who provided interesting insight and described important work in research that was seemingly done independently from government.
However, it is exceptionally difficult to separate science from politics or impacts therefrom. Very little science is done without money. Politics are intimately involved in its allocation (money). Therefore, politics certainly are involved and may be even overarching.
Are you channeling President Eisenhower?
That’s right. Rummo may be trying to be subtle in not naming names, but it comes off as weasel words that can be read equally as an attack on Trump or at least as a dismissal of his efforts to speed up the wheels of plodding government bureaucracy. I hope that that was not Rummo’s intent.
Democratic activists and journalists (oops sorry for the redundancy) will of course only see that as an attack on Trump.
To my way of thinking, taking actions to improve a situation, even if those actions are motivated by political considerations, and even if blatantly trumpeted for political reasons, is a totally different class of offense than sniping from the sidelines at the positive efforts for purely destructive political motives. The latter is what presidential candidate Harris and Trojan Horse surrogate Biden have done repeatedly.
Actually just the opposite. The intent was primarily to spotlight science not politics and secondarily as a critique of those almost exclusively democratic leaders who have maintained they would not accept an approval from the FDA because they have ignored all the science up to this point and somehow equate an EUA with President Trump pressuring for a recklessly rushed approval.
Thanks for clarifying
I doubt a vaccine will help much at all honestly. This is a shamdemic based on a spurious test for a large part of a virus that mostly isn’t that bad. If we are going to shut society down for things like this then might I suggest we enact the following too? Ban on driving – road deaths, ban on smoking anything or vaping – cancer, ban on swimming – drownings, ban on flying – potential for accident. In fact, let’s not ever leave our homes again, wrap up in bubblewrap and pray we don’t get food poisoning.
Meanwhile, can someone tell me why the vaccine isnt dead COVID-19 viruses like the flu vaccination I had on Monday? I would really like to know.
“…why the vaccine isnt dead COVID-19 viruses…”
Because somebody wants to make billions selling a drug (not a vaccine) that alters peoples DNA, and doesn’t want to have mid to long term effects discovered.
It kinda reminds me of Thalidomide. My mother was offered Thalidomide while pregnant with me, but her common wisdom was to not take anything during pregnancy. Thank you Mom.
G posted: “This is a shamdemic based on a spurious test for a large part of a virus that mostly isn’t that bad.”
Hmmmm . . . over 1.1 million people worldwide, and over 220,000 just in the US, whose deaths are attributed to COVID-19 (with or without comorbidities)? And some initialed person (bot?) has the hutzpah to post “a virus that mostly isn’t that bad”??? Go figure.
CDC says that about 6% of the deaths were “without comorbidities”. That means about 13,000 deaths due to the virus alone. “Mostly isn’t that bad” is a pretty accurate statement, considering that close to 3 million people die each year in the US.
Another way to say that 0.4% deaths are caused by COVID-19.
The more important number we need to know, is the total mortality curve for this year in a comparison with 2015, 2016, 2017, 2018 and 2019.
This would be the only graph giving a bit of meaning. – Why is this so seldom mentioned?
Excellent point about excess deaths, Carl.
Does anyone have a link to the data on that?
RD, the US weekly data is publicly available. Google cdc excess deaths. Takes you to their configurable dashboard.
Thanks Rud
For all US, total excess deaths since 2/1/2020 are estimated by CDC to be 231,952 – 311,882 (as of 10/22/2020).
If we can trust the numbers, it looks like roughly 10x worse than a flu season. Only about 10% of it in NYC
The best I could find, regarding mortality 2015 to 2020, was:
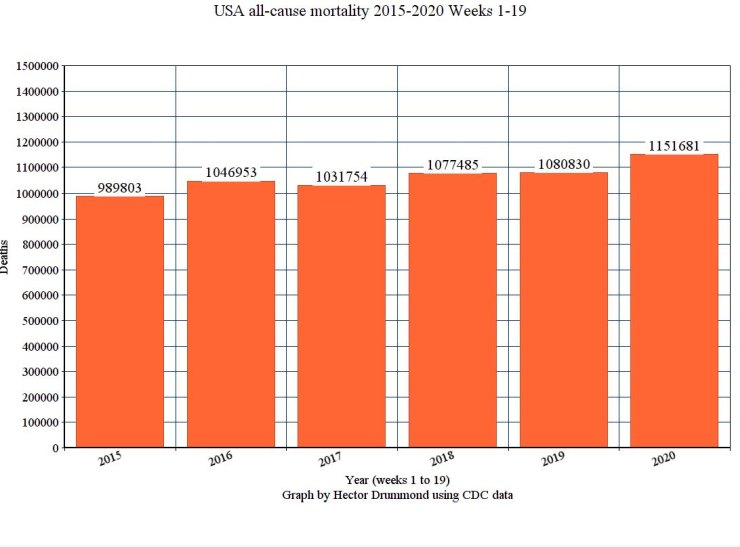
You will still have to adjust for population increase. But, the difference between 2019 and 2020 so far is: 1151681-1080830 / 1151681 * 100 ~ 6% increase.
All other graphs are incorporating influenza, COVID-19,etc dependencies.
Carl,
I’m not vouching for the CDC’s methodology or the accuracy of their raw data, but they claim to base the expected deaths on the “historical trends (from 2013 to present) to determine whether the number of deaths in recent weeks was significantly higher than expected, using Farrington surveillance algorithms…”
If they are ginning up fake deaths, we’re in a worse situation than I have imagined.
Remember that these numbers are deaths from all causes. It doesn’t involve any attribution of causes, so it doesn’t matter if every skydiving accident and suicide was mis-coded as a Covid death in order to scam money from the government.
Unless they have faked the total number of dead bodies, or their “Farrington surveillance algorithms” are yielding a bogus number of expected total deaths, excess deaths are way above any normal flu season.
Given what I have seen in the past few months, I am only about 80% confident that the actual death numbers are accurate, and maybe 50% confident that the expected deaths model is reasonably legit.
But let me hasten to add, I am no expert.
Thanks Rich Davis.
It is really confusing. The Swedes get their data from the “taxman”, which I am more confident with.
I downloaded the spreadsheet from:
https://gis.cdc.gov/grasp/fluview/mortality.html
using column D and K, created a graph and realized the sheet is so disorganized or “advanced” that you have to reorganize the data, before you can make a graph.
From what I saw, 2020 surely look like a bad Spring and fine over the last months.
Hiskorr, why do you conclude that a having a comorbidity necessarily rules out COVID-19 as a cause of death?
If I have a cancer that is going to kill me in ten years and COVID-19 is the predominate cause of my death within 3 months, should I excuse COVID-19 because I had terminal cancer?
The 220,000 number is certainly inflated (i.e., doesn’t reflect reality), but even with that inflated number the chance of dying if less than 70 y/o is 0.5% (i.e., 99.5% survival). Most who die are those at their end of their lives and/or those with severe co-morbidities (including being intubated). In other words, it is an illness that picks off those with a very low life expectancy. That qualifies as a generally not dangerous illness.
I have to agree. There are a number of factors that are being ignored regarding deaths ’caused’ by Covid-19. Since a large number of those deaths have been taking place in long-term care facilities, and many residents of those LTC facilities have co-morbidities and, as my missus keeps reminding me, many of the patients there aren’t there to get better but for extended end-of-life care, then the question has to be asked “How many of those Covid-19 deaths had nothing to do with Covid-19?” So someone who has terminal Stage 4 cancer and is days from dying but also tests positive for Covid-19 is considered a Covid-19 death when they finally pass even though they may have had no symptoms of coronavirus?
Yes, this is an extreme case, but any number of co-morbidities could have been the actual cause of death while Covid-19 contributed little or nothing to their deaths. I haven’t seen a breakdown of the co-morbidities involved or their contribution to death.
Perhaps one way to look at this would be to track the excess number of deaths as compared to what is normal for a given population. The case might be made that these excess deaths have a connection to Covid-19, either directly through infection or indirectly due to other life-preserving treatments/cures not being available for those with other health conditions due to the shutdown of medical care facilities because of Covid-19. But for sake of argument, let us assume deaths caused by the lack of health care due to shutdowns is nil.
If the normal number of deaths not caused by accidents/misadventure/homicide for a given population over a six-month period is ‘X’, but the number of those deaths that take place is actually X+3%, and that 3% is outside the standard deviation (let’s use 1% for the standard deviation for this example) and changes in population (in-/out-migration, seasonal variations) have been accounted for, might that excess 2% be caused by Covid-19?
DCE
Please see my comments above. The excess deaths number as calculated by CDC, is higher than the deaths attributed to Covid.
Seems a legitimate question to ask, whether lockdowns or any other special cause may have contributed to the total deaths in a significant number, relative to deaths caused by, or hastened by covid.
In other words, the overall effect of the pandemic and our response to it (and any other unusual situations that we haven’t considered), has been 232k-312k excess deaths since Feb 1st. (With caveats I gave above). It’s apparently not the case that we’ve seen roughly the normal number of deaths per million. There is a real, large excess from some cause, that can’t be attributed to non-covid deaths being mis-coded to game the reimbursement systems.
Why don’t we shut society down because of deaths from road accidents, smoking, cancer, drowning, flying etc. Maybe because those things aren’t infectious; we can’t catch them by social contact? The one exception in that list is smoking. We do have social controls, such as non-smoking venues, to limit contact between smokers and non-smoker to reduce the risk to non-smokers of ‘catching’ the negative health effects of smoking.
If you don’t take action then you don’t have only 3,000,000 cases in US you have a cumulative total of perhaps 50 to 65% of the population for herd immunity i.e.170,000,000 cases allow half those to be naturally immune and you still have 80,000,000 sick people.
And herd immunity only works if catching the disease gives continued immunity – no proof yet
It is unwise to let it run its course.
ghalfrunt
https://scitechdaily.com/long-lived-antibodies-detected-in-both-blood-and-saliva-of-patients-with-covid-19/
The Swedish approach was never to “let it run its course”.
The Swedish approach was to have confidence in the common sense of their people to appropriately practice social distancing on a voluntary basis, as appropriate to each individual’s risk factors, without crippling their economy, and focusing protections on care homes (nursing homes) where the risk was greatest.
There is a lower population density and a lower average household size in Sweden, than in many other countries, so this approach may not have worked as well in London, or New York. On the other hand, no approach could possibly be as bad as forcing facilities full of the most vulnerable population to accept infected patients. That approach k!lled more people in NYC than any other factor. How could you make worse policy choices if you were trying?
HIT (herd immunity threshold) is based on the idea that if enough people are immune, the virus cannot spread. Contrary to what you say, it is not inevitable that everyone eventually gets it. An analogy could be made to fire throwing sparks into an area soaked with water. Unless the spark is able to ignite something, it will die out and the fire will burn itself out. HIT for covid may be less than 20% due to the population having prior exposure to similar corona viruses.
We didn’t know all this in February and it was not unreasonable to take the lockdown approach initially. KIt was a brave decision made in Sweden. We should learn from our failures and from their successes. Once the election is over, maybe we will.
The Covid-19 crisis will vanish without a trace – or an explanation – if Democrats sweep the election.
The Swedish government is not allowed to declare a state of emergency in peacetime. Thus, the main factor behind Swedish exceptionalism during the present pandemic is that the Swedish constitution prohibits the use of lockdowns as seen from Chapter 2, Article 8 in the Swedish constitution (Regeringsformen).
“Everyone shall be protected in their relations with the public institutions against deprivations of personal liberty. All Swedish citizens shall also in other respects be guaranteed freedom of movement within the Realm and freedom to depart the Realm.”
That’s interesting Robert. Imagine a government that actually thinks the constitution is more than an aspirational document! Isn’t the Swedish constitution a living document? How backward! Seems that they need to get busy packing their courts.
oddly(not) ALL the early reports on smokers getting Covid LESS than non smokers has been dropped?
smokers who did get it seemed to have a 50/50 of death but again age related as well from the dailymail UK reports that did get some reportage
So that is why president Trump said the other day, there would be a vaccine before the end of the year.
This is good news, although I feel it would be even better to make promotion for good diet and free consultancy with your doctor to evaluate Your specific best way to improve your health.
Secondly, we need to stop Alphabet from censuring advice from American Frontline Doctors and similar competent persons, unions and organizations.
Anyway, thanks for the post Gregory J. Rummo
From the above article: “Drug development begins with a conceptual design model followed by research, engineering, small scale manufacturing and several phases of testing . . .”
Actually, that statement is a little too simplified. 99+% of drug development begins, first and foremost, with the commitment to, and release of, MONEY to fund the development. And who is the biggest possible funder in the world: the US government.
That is why, to date, the US government has spent/committed over $6.5 billion dollars amongst 7 drug development and manufacturing companies to kick off and/or accelerate the development of COVID-19 vaccines, with another $2.5 billion allocated just for vials to store the vaccines, syringes to deliver them, and increased manufacturing capacity. (ref: https://www.usatoday.com/story/news/health/2020/08/08/feds-spending-more-than-9-billion-covid-19-vaccine-candidates/5575206002/ )
If they do manufacture a vaccine that is safe, effective and widely available at a reasonable cost, the political and healthcare bureaucracy will have a way to end the mess they’ve created with their panicked response to the virus, and save face at the same time.
How do you prove a vaccine is effective ?
Seems impossible to me.
Stop the hysteria. This is NOT polio.
NOBODY dies solely from covid 19
Here in Denmark, we have the lowest mortality rates seen in 5 years !
In a double blind study – so that there is no bias – participants are closely monitored for Covid19 infection post vaccination. If a statistically significant cohort of vaccine recipients remain healthy while the placebo cohort becomes infected then you have demonstrated efficacy.
One vaccine is undergoing challenge trials. Volunteers are vaccinated then given a controlled dose of the ‘rona.
IMO, this is better than RCT. RCT should still be used, but the challenge trial should give us a better understanding of the real effectiveness of the vax.
N. Jensen,
You posted: “NOBODY dies solely from covid 19.”
Do you have any facts . . . any smidgen of objective data . . . to support that dogmatic assertion?
I will not waste my time waiting for your reply.
One of the test persons of AstraZeneca Oxford vaccine died yesterday on corona.
Well, some say he got placebo, but I haven’t seen an official statement from Oxford yet.
The guy was 28, young and healthy
Remember, there still no effective vaccine against SARS/MERS, because the vaccination results in an unpredictable immune response and death when exposed to the virus.
If he did get the placebo it was a meningitis vaccine that has the rare side effect of anaphylaxis, which can lead to death.
He was apparently given the placebo so not covid vaccine related
He was doctor treating CV-19 patients in Brazil. The Brazilian news story was he took the placebo and AstraZeneca said they couldn’t disclose whether he was on the placebo regimen or vaccine regimen; however, they did report they had no safety concerns from the death and the study will continue.
If he was on the placebo, AstraZeneca’s actions corroborate it even without actual confirmation.
That is wrong, (I am pretty sure), he didn’t contract covid or die from that. He died from effects of the placebo.
But what sense does it make to give a “placebo” with active ingredients? Why would you not produce a placebo from a saline solution and if necessary to maintain the double-blind protocol, add harmless ingredients to make the placebo look the same as the actual vaccine?
Something is fishy here.
News report was that he was given a meningitis vaccine. Perhaps they were doing parallel trials – with the COVID placebo group getting a new meningitis vaccine and the meningitis placebo being the COVID-19 vaccine. I am not sure that is a sensible approach to all but beancounters.
Vaccines are designated as biologics not drugs and don’t require the same amount of safety testing.
NO LIABILITY: since the passing of the vaccine injury compensation act, drug manufacturers have been shielded from lawsuits by customers injured by their vaccines. This significantly increases the value of vaccines because there is no cost for legal defense or settlements like the 4.85 billion settlement against MERCK for its drug VIOXX.
NO LONG TERM/PLACEBO SAFETY TESTING: Vaccines are not required to go through long term safety studies with real salt water-based placebos.
In terms of safety studies, a major issue is that most vaccine studies use another vaccine as the control placebo, or use the background substance of the vaccine. There is only one recent study (Cowling 2012) where a true saline placebo was used, rather than another vaccine or the carrier fluid containing everything except the main antigen. That study showed no difference in influenza viral infection between groups but astonishingly it revealed a 5-6 times higher rate of non-influenza viral infections in the vaccinated. It is no small wonder more true placebos are not used in vaccine research. (Source: drsuzanne.net)
The lack of adequate testing makes vaccines cheaper than drugs to take from trials to market. Furthermore, it is becoming increasingly more expensive to do the long-term safety studies on drugs because the test group cannot be using any other drugs at the time of testing. With over 70% of the US population using prescription drugs this becomes nearly impossible to find. (Source: CBSNews.com)
Cytokine cascade? That can affect someone of any age but is far more prevalent in younger adults. That’s what killed so many during the Spanish Flu in 1918, and it killed quickly. (The saying back then was that it killed so quickly that you’d “have breakfast with your family and supper with God.”) That someone might have the same reaction to a vaccine isn’t surprising.
Soon is not soon enough. Given how the virus spreads now with sinking temperatures, it is only a matter of weeks till everyone is going to get infected. And we have 6 months of winter ahead, at least in Europe.
“…it is only a matter of weeks till everyone is going to get infected.”
Of course in Sweden, they have decreasing deaths and cases (from having reached HIT?), have no need for a vaccine and had almost no lockdown or mask wearing from day one. They acheived this by letting young people get the virus while their immune systems were strongest.
The rest of the world has taken the wrong road.
Yes, there are two paths you can go by, but in the long run, there’s still time to change the road you’re on.
And it makes me wonder!
Stop leading the pack astray leitwolf. Listen to Timo.
I’m a bit suspicious about winter spread.
Here in Australia we have just gone through winter and no uptake in the virus contagion caused by the weather.
The Southern Australian state of Victorian suffered a blow out in cases but this was due to bureaucratic bungling. e.g. Hiring low paid security staff . Security staff that were double and triple shifting jobs.
One shift guarding quarantined suspect virus positive people in hotels, The next shift working in aged care centers and the next shift working at supermarkets. Not to mention going home and not self isolating themselves away from their friends and extended families.
Some statistics.
According to a study done by the University of Wollongong (NSW). Median age of Covid 19 IFR fatalities in Australia is 82years.
Total deaths from Covid in Australia so far – 905. Far less than the average flu season and way less than a serious flu season. On the other hand, because of Covid lockdowns, social distancing, general health habits and masks, this years flu deaths number as a handful.
Politics funded the vaccine search by guaranteeing sales whether they are effective or even needed and used.
Work on a vaccine and we will buy a guaranteed 100,000,000 doses from you, even if you fail, even if no one wants to take it.
Is that supposed to imply that it’s a bad thing?
Yeah, right. They should have only funded the one that was going to work best. And they shouldn’t have wasted all that money making ventilators that weren’t actually used.
If only the virus had waited for President Harris to be in office! Then the smart decisions would have been made to avoid waste.
It’s not at all clear that the bureaucracies involved want to “end the mess they’ve created” and relinquish the power they’ve accumulated. Nor is it likely that the WHO, CDC, and FDA, in particular, will be able to “save face”.
shame on those politicians
Who, er, know no shame.
This is just weird.
Does no one realise that throwing money at a vaccine or mab will make safety testing quicker. It reduces funding delays, it can allow drugs to be produced in the hope that they will be safe and effective (used or perhaps thrown away depending on testing).
It will not allow the long term safety to be speeded. (birth defects, cancer, etc take time to develop)
The UK vaccine is to start “human challenge” studies where up to 90 healthy people will be deliberately exposed to Covid.
The trials, which could begin in January, aim to speed up the race to get a Covid-19 vaccine.
Results in May 2021!!
Human challenge volunteers get paid £4,000
If the volunteers being paid to test the vaccine by exposure to COVID are young and healthy the conclusion will be that the vaccine is useful as they will all pass the disease asymptomatic or mild, as they would if they are not vaccinated.
Volunteers should all be >75 years old and healthy, and should be paid quite a lot more. Let’s see how many die despite the vaccine. I would certainly not like to be in the control group of that test that is given the placebo and marched against the virus.
The AstraZeneca vaccine candidate is one of the few potential SARS-2 vaccines that I think is a viable candidate.
Agree.
IMO mRNA vaccines are dangerous.
There is the problem that Big Pharma and WHO have interests against those of the population. Big Pharma is after the business of selling vaccines whether necessary or not, and WHO officials bribed by Big Pharma play the alarmist card.
The 2009 swine flu (H1N1) flu pandemic had very little danger with a lethality of only 0.01-0.03 %, yet the WHO and Big Pharma so overplayed its danger that many countries bought huge amounts of vaccines that were never used. Spain bought 13 million doses (47 million people) at a cost of 98 million € (over 100 million $). Only 3 million people showed up to be vaccinated as the population distrusted a rushed vaccine and the reports showed the 2009 flu pandemic not more dangerous than seasonal flu. 4 million doses were donated and the rest destroyed.
https://www.20minutos.es/noticia/787110/0/destruccion/vacuna/gripe-a/?autoref=true (in Spanish).
Following the advice of Big Pharma or the WHO may not be in the best interest of the population. The vaccines against COVID are being rushed and there are lots of economic and political interests behind them. It is not a question of trusting the science. As we saw with hydrochloroquine, science is just another battlefield for money and power. The problem is that we can no longer trust the institutions, like WHO or our governments to defend our interests.
The problem is that we can no longer trust the institutions, like WHO or our governments to defend our interests.
That’s for sure. Big Pharma also influences media coverage with huge ad buys, lobbies politicians, and works to destroy and delegitimize opposing views, following the climate change template.
Very well said Javier “As we saw, science is just another battlefield for money and power. The problem is that we can no longer trust the institutions, like WHO or our governments to defend our interests” !!
Who will look after the interest of common people? Now people like you step up and raise your voice… Sensible people need to step up urgently! Otherwise, those people with greed can never be stopped.
UK plan to be first to run human challenge Covid trials
https://www.bbc.co.uk/news/health-54612293
After exposure to Covid, the young volunteers will need to stay in a biosecure facility until they are no longer infectious.
They will be financially reimbursed for their time, and monitored for up to a year after taking part in the study to check for any side-effects.
People can sign up here.
Purposely infecting someone with Covid does pose an ethical dilemma, especially since there is no treatment to cure patients, although there are ones that might make it less deadly.
Prof Julian Savulescu, an expert in ethics at Oxford University, said the trials were justified: “In a pandemic, time is lives. So far, over a million people have died.
“There is a moral imperative to develop to a safe and effective vaccine – and to do so as quickly as possible.
“Given the stakes, it is unethical not to do challenge studies.”
“So far, over a million people have died.” = .0013% of world population US deaths = .07% Highest deaths by country = Peru at .1% This is a media driven pandemic (no intent to diminish the tragedy).
Young volunteers won’t give them the answer. They need old volunteers.
Infecting them is a big ethical and legal problem, but you can let them infect themselves. Boxers hit each other in the head all the time and it is known to have terrible effects on their health. Yet it is not a problem because they know it and they do it to themselves. And they also do it for the money they get paid.
Anyway as soon as they start distributing the vaccines they will get scores of unpaid volunteers. In China and Russia they are already vaccinating a lot of people that won’t get a dime. Me, I’ll wait and see.
Javier October 22, 2020 at 9:34 am
Young volunteers won’t give them the answer. They need old volunteers
——————
NO
If the vaccine is effective then the volunteers receiving the vaccine should show only mild or no symptoms. Whilst those receiving placebo (is this happening this time) should show worse symptoms
This is done to volunteers because it is not really ethical. Giving it to oldies is murder.
Young people are already showing only mild or no symptoms without vaccine.
Stop with your nonsense Ghalfrunt and stop telling people to drink bleach. Shame.
My modest proposal is to require all federal, state and local politicians be vaccinated 4 weeks before the general public. Same with every member of the healthcare bureaucracy, every nurse and doctor, all judges and lawyers, and every member of the MSM. No exceptions.
If there are no “complications” we’ll know the vaccine is safe for the rest of us.
That should include every employee of the company that produced the vaccine, too.
The reason for having to do challenge studies is that there is not enough virus around to get statistical significance.
President Trump, Dr Zev Zelenko and the Front Line Doctors would disagree that there is no treatment. If given early on known contact or on first symptoms a treatment to bolster the innate immune system works at close to 100% efficacy. This is as a minimum: zinc, zinc ionophore, antibiotic. Intracellular zinc blocks RNA viruses from replication using the cells’ RNA transcription mechanism. This innate immune system function of zinc has been known to block corona viruses, influenza viruses and polio viruses. [ DOI: 10.1371/journal.ppat.1001176 – Published: November 4, 2010]
Had these venal medical bureaucrats told everyone to be sufficient in Vitamin D and Zinc – thousands of lives would have been saved worldwide – and there would have been no pandemic. Not only that influenza and polio would also be significantly reduced – which is why the medical bureaucrats kept quiet and instead repeated the untrue mantra of no normality until there is a vaccine.
Right, Ian. Not to mention the other treatments that have proved to be effective. You won’t get any of that in a hospital unless you are POTUS. My friend sat right here beside me at the computer for an hour or more. Three days later he tested positive for covid. A week later he was airlifted to the covid unit. A month later he was gone. He was given remdesivir, which we know does little or nothing, and something else, he wasn’t sure what. Oxygen mask 24/7 towards the end and then intubated. Could any of the treatments not approved by the FDA have saved him? We’ll never know. (b***ards)
promises, promises….I hope he’s right but I watched a vid last night, equally enthusiastic, about how recent breakthroughs meant that break even for fusion was only 2 or 3 years away. Turned out the vid was 5 years old…..
Yet every year the flu vaccine can be rushed without anyone complaining…
Funny thing, if true, would be the worlds first vaccine for a corona virus.
World’s first mRNA vaccine for a Corona virus. There are CV vaccines in use for quite some time on the animal health side of PHARMA.
yes but the animal corona is a gut version, and from my vet boyfriend is better then nothing but not all that great either. havent sussed it out but Id guess? its a killed virus?
https://www.merck-animal-health-usa.com/nobivac/nobivac-canine-1-dappvcv
yup merks at least is an inactivated
wouldnt use that on my dogs as the combo has live parvo not killed
Avian coronavirus vaccine is against bronchitis.
…would be the worlds first vaccine for a corona virus.
…and prosperity and fusion are right around the corner.
I don’t understand something and perhaps someone can help me out here. I read recently that the Phase 3 readouts weren’t until 2022 late or early 2023. They are double blind so no data can be before they read out. And that the results that the FDA are deciding on will be very small Phase 2 data. Is this correct?
Phase 3 trials are 2 years. I received the first injection on Aug 1 and the second 28 days later. Eight vials of blood have been drawn now three times, on Day 1 then after each injection. Moderna knows by now what the levels of neutralizing antibodies are in vaccinated participants vs. those who received placebos. But they want to know for how long they persist. That is a longer term goal but if it is shown that antibodies persist for at least three months, that would be enough to grant an EUA assuming safety while the monitoring phase of the study continues.
“What is currently known about the two leading candidates’ mRNA vaccines—Pfizer and Moderna Inc.—is that they are showing safety and efficacy in late stages of phase 3 clinical trials.”
The Phase 3 end point is only something like when about 30-35 sham vaccine recipients have gotten COVID-19. Then that very low number will be compared to the COVID-19 number in the actual vaccinee group to see if it is significantly different. That is how efficacy will be measured. That vaccine efficacy story is only part of the overall safety story of the vaccine though.
Simply noting whether or not the vaccine itself (before the vaccinee is exposed to the live virus in the community) causes safety issues is only half the story. I have no doubt these mRNA and the DNA vaccines can induce an IgG antibody response after the booster. But the safety and efficacy will not be fully addressed until hundreds of vaccinees have actually been exposed the SARS-CoV-2 some 6 to 12 months after the booster when IgG levels are starting to wane. Simply inducing an antibody response to a respiratory viral pathogen is quite problematic to T-cell immunologists who understands how priming the immune system for this class of pathogens is critically dependent on a robust Th1 T-cell memory response induction. A misprogrammed immune response by trying to fool the immune system with either mRNA/DNA vaccine or an inactivated virus vaccine is likely to happen in some small percentage of the vaccine recipients. Trying to fool the immune system can easily cause problems for some people when the real virus is encountered.
I doubt Greg Lummo, lecturer in Chemistry, fully understands the T-cell immunology and T-cell memory recall issues involved here. Issues like how an Antibody Dependent Enhancement (ADE) pathology may occur some 6 to 24 months from now in the vaccinees who become infected with SARS-CoV-2 with a waning antibody titer that is not fully neutralizing for the virus without a robust T-cell Th1 memory response to eliminate infected cells.
Or how an asthma-like eosinophilia may be induced by a RNA/DNA vaccine. Induction of a Th2 memory response to a respiratory virus pathogen may lead to Airway Hyper-Reactive (AHR) in both the upper bronchioles and deeper in the lung epithelium. This is classically caused by a Th2-programmed cytokine storm led by Interleukin-5 (Il-5) and eotaxin from pathogenic Th-2 CD4+ T-cells that drives this asthma-like response.
Accordingly, these Moderna and J&J mRNA modality and the DNA modality vaccines are likely dangerous for some people IMO.
=========
As a completely separate issue, I have seen where both Gov Newsom of Cal and Gov Cuomo of NY are questioning a vaccine. This is of course being politically-driven by them to damage Donald Trump. What they also have come to relish is the power rush these two men have gotten from ordering the lockdowns and the dictator-like satisfaction and attention on them it delivers via dopamine signaling deep in the brain. So both these Democrats are high on power, it’s an opiate-like rush to the brain. Neither obviously wants to surrender this new found power they have to lockdown their residents and order businesses around like petty little Banana Republic dictators. Thus Newsom and Cuomo don’t want a vaccine anytime soon and end their power addiction high.
Great post. I assume from your post you understand that this mRNA vaccine finds it way to the ribosomes of cells and translates the spike protein which the immune system recognizes as foreign and produces antibodies against. I have heard of no severe adverse reactions in Phase 3 which has a cohort of older people (+65 yo). There were adverse reactions during an earlier phase study; myalgia, injection site soreness and fevers but with the higher dosages. Scott Gottlieb has said that an effective vaccine, even if immunity is relatively short lived, coupled with herd immunity would be sufficient to end this pandemic. I hope he’s right.
Yes. But simply inducing an IgG antibody response is child’s play immunologically speaking. That is what the mRNA and DNA modalities are doing. It is naively foolish in my opinion. (I my doctoral work was specifically in human immunology-virology where I focused on CD4+ T-cell memory recall immune responses to acute respiratory viral pathogens.)
Protecting the fully naive to coronaviruses host against this nasty SARS-2 respiratory virus, an immune response that requires a properly programmed T-cell response, is not going to be properly programmed by a mRNA or DNA vaccine. In my estimation, some people who are asthmatics or prone to autoimmunity will be worse off getting the mRNA or DNA modality vaccine.
All of our primary immune inducing vaccines to respiratory associated viral pathogen, from MMR vaccine to Smallpox vaccine to the Yellow Fever, are live, attenuated virus vaccines. There is a reason for that. A very good reason. One that apparently not enough immunologists working in Big Pharma or the US government understand.
Even assuming late stage approval occurs – the issue of manufacture, distribution and administration of said vaccine is still extremely problematic.
The head of the largest vaccine maker in the world (located in India) said the industry doesn’t have the capacity to manufacture enough vaccine until 2024.
Plus, it seems the vaccines in the US are 2 stage and are also highly environmentally sensitive (i.e. must be stored essentially frozen).
All this, plus the serious anti-vax movement both among far right and “natural” left, means the likelihood of a national herd immunity via vaccine, in the next year or less, is close to zero. It is likely to be much longer.
c1ue…not close to zero, but zero. If what I’m reading is true.
“It is a common misconception that an approved vaccine will provide “silver bullet” immunity, a scenario based more on a Hollywood film narrative than reality because no Covid vaccine trial protocol defines its “success” as:
– Providing immunity from infection from the SARS-COV-2 virus
– Reducing mortality risk from the COVID-19 disease
– Providing immunity from COVID-19 disease symptoms
Instead trial “success” is defined as an amelioration of COVID-19 symptoms in 50-60% of volunteers, who are healthy adults likely to be at risk only from a mild or asymptomatic infection and thus not even a population group facing significant mortality risk from COVID-19. These dud Covid vaccines aspire to be buckshot not silver bullets…”
——————–
Early data from the Oxford study, for example, suggest the vaccine may not provide complete protection – perhaps only warding off Covid-19’s worst symptoms.
“Ideally we’d like immunity from a vaccine to completely wipe out a virus,” says Dr Andrew Preston of the University of Bath. “But if we can reduce Covid-19 to a seasonal cold, that would be good enough compared to the current alternative.”
“[The latest research suggests] there is no guarantee that a vaccine will be found that confers lasting immunity,” adds Stephen Evans, professor of pharmacoepidemiology at the London School of Hygiene and Tropical Medicine. “It’s possible immunity will be short-lived or even non-existent.
telegraph.co.uk
14 July 2020
i’ll never take any vaccine
i’ll take my chances
i’ll never wear a mask
i am not afraid of living.
i’ll never wear a mask
The stores I shop at say, “No Mask, No Service”
I pretty much don’t have a choice.
oronavirus vaccine volunteer in Brazil’s AstraZeneca trial dies — but authorities say trial to continue
Sounds shocking until you read that the death was a volunteer in the “placebo” group.
Who knew placebos could be so dangerous ?
Getting a Vaccines is an interesting risk play. Say there is small risk of it causing you problems, this has to be weighed against risk of catching the disease it is suppose to prevent. But another angle is even if you don’t get the vaccine but enough other people get the vaccine your risk is also reduced of getting the disease. For example say you are sure 90 pct of people will get it, in that case best play for you would probably not to get vaccine. But if enough people think that way then there will not be enough people getting vaccine to stop the spread.